

How to Cite | Publication History | PlumX Article Matrix
In–Silico Visualization of Gene-Gene Interactions in Autism Spectrum Disorder Genes
Abhinob Baruah, Kajal Singla, Shilpa S Chapadgaonkar and Rashmi Rameshwari
Department of Biotechnology, Manav Rachna International Institute of Research and Studies, Faridabad, India.
Corresponding Author E-mail: rashmi.fet@mriu.edu.in
DOI : http://dx.doi.org/10.13005/bbra/2852

ABSTRACT: Autism spectrum disorder is a group of neurodevelopmental disorders with still unknown causative mechanisms. Recent findings implicate a complex etiology with multiple genetic and epigenetic factors. The disruption of vital gene-gene interactions has been proposed to be one of the contributing factors for the disease. NPAS (neuronal PAS domain proteins 1 and 3) have been recognized as critical regulators of neuropsychiatric development and function. In the present study, a genetic network association with NPAS3 and the highest interacting gene CRKL has been mapped for ASD and analyzed to decipher the complex genetic basis of Autism spectrum disorder. CRKL encodes a protein kinase with SH2 and SH3 (SRC homology) domains which activate RAS and Jun signaling pathways. The CRKL gene sequence was retrieved and compared using BLAST homology search. The physiological and chemical features of CRKL protein was studied using Uniprot Database and Protparam followed by homology modelling by constructing a phylogenetic. Physio-chemical properties of CRKL protein was studied by Protparam which gave insight into the role of other interacting proteins involved in this process. Further analysis, a conserved region of intersecting protein of CRKL were identified using multiple sequence alignment. The gene interactions data revealed that CRKL as one of the key genes associated with Autism spectrum disorder. CRKL defects in the deleted region of 22q11. 2 of the human chromosome 22 have a strong correlation with several of the birth defects as it can hamper the development of the brain along with behavior and cognitive function. This may lead to neurodevelopmental disorders like autism spectrum disorder or schizophrenia, depending on which particular region in the gene is affected. CRKL gene could be a potential biomarker for many In-borne diseases. An investigation in current study was done to find out the genetic network and decipher the complex genetic basis of Autism spectrum disorder.
KEYWORDS: Autism Spectrum Disorder; Biological Network Visualization; CRKL; Gene-Gene Interactions; RAS/MAPK
Download this article as:| Copy the following to cite this article: Baruah A, Singla K, Chapadgaonkar S. S, Rameshwari R. In–Silico Visualization of Gene-Gene Interactions in Autism Spectrum Disorder Genes. Biosci Biotech Res Asia 2020;17(3). |
| Copy the following to cite this URL: Baruah A, Singla K, Chapadgaonkar S. S, Rameshwari R. In–Silico Visualization of Gene-Gene Interactions in Autism Spectrum Disorder Genes. Biosci Biotech Res Asia 2020;17(3). Available from: https://bit.ly/3iS2ooP |
Introduction
Autism spectrum disorder (ASD) are known to have an exceedingly complex multigenic neuro-developmental disorder. It manifests as impairments in growth and differentiation of central nervous system which ultimately results in difficulty in speech, communication and social behavior, abnormal response to stimuli, poor eye contact, and repetitive behavior in kids (Bonnet-Brilhault 2017; Hagerman, Rivera, and Hagerman 2008; Kaufmann et al. 2004; Rogers, Wehner, and Hagerman 2001). Autism spectrum disorder shows prevalence of 1 in 68 children affecting male more than females (1 in 42 boys and 1 in 189 girls). Male to female gender ratio came out to be 4:1, as reported for Autism spectrum disorder (Fombonne 2009). The phenotypic variations in Autism spectrum disorder are inherited unlike the environmental factors (Abrahams and Geschwind 2008). It has been reported to be associated with parental hereditary factors, stress, improper uterine environment and other biological causes. These factors have also been correlated with various other disorders such as schizophrenia, fragile X syndrome and mitochondrial disorders.
Several genomic studies were conducted worldwide on the autistic families to discover the genetic association of ASD (Beaudet and Arthur 2007). The studies highlight the baffling genetic as well as phenotypic heterogeneity in ASD. Genome-wide association studies involve screening to identify deletions, duplications, CNV (copy number variations) and SNPs (Single Nucleotide Polymorphism) that could be correlated with ASD phenotype. Genetic linkage and mutation studies have reported multiple mutations responsible for ASD (Sykes and Lamb 2007). This was seen in a great number of autistic individuals accompanied with their unaffected family members. It could have been a consequence of a sum of variations and impetuous changes in the genetic material during meiosis (Beaudet 2007; Gai et al. 2012). Non-syndromic autism has been shown to be connected to a large number of earlier unknown rare mutations and gene number variations. This was made possible through extensive Genome-wide association studies and other genetic analyses which have been instrumental in throwing light on various associations between these mutations and the pathology. However, the complete mechanisms and cure still remain elusive to the scientific world.
Previous studies indicated a significant amplification in ASD as seen in Mendelian disorders of the Ras/MAPK pathway or (RASopathies). They have all the upregulated typical symptoms of ASD (Zuk et al. 2012) (Consortium and The International Schizophrenia Consortium 2009). In a recent study, RASopathy gene SNPs testified for presence in the Autism Spectrum Disorder patient with GWAS through various pairwise interlinkages of genes that met the standards of genome wide criteria for significance (Shihab, Dawood, and Kashmar 2020). Lastly, an orthogonal approach provided confirmation for the influence of epistasis in Autism spectrum disorder which revealed that the Ras/MAPK pathway plays a pivotal role in idiopathic Autism Spectrum Disorder (Mitra et al. 2017).
Receptor tyrosine kinase is an enzyme responsible for transmitting signals from adjacent cells, particularly activate RAS activity. This successively promotes rapid events leading to energize one or more isoforms of the RAF kinase, accompanied by MEK and ERK kinases activation and, eventually, the expression of target genes. Naturally, this ‘linear’ pathway is submerged in a web of other pathways, as well as enzymes such as mTOR and PI3K. As such, changes in the activity of this matrix affect the product of the ‘canonical’ or standard, RAS pathway. Mutation in the network is an elementary syndromic form of autism, the first suggestive evidence showing that the RAS pathway might be essential in autism.
Recently, string of discoveries has shown that ASD is distinguished by mutations in various genes in the RAS pathway, each of which leads to a gain-of-function of ERK signaling their review of these disorders. Gary Landreth and his fellow workers observed that the collection of phenotypes linked with 16p11.2 deletion — obstruction in the central nervous system, heart and brain, along with facial dysmorphology — is also seen in the RASopathies, comprising DiGeorge syndrome that is caused by the 22q11.2 chromosomal region 4 deletions. If 16p11.2 deletion results in a RASopathy, then one would say the classical RASopathies to demonstrate a high liability to autism (Samuels, Saitta, and Landreth 2009).
Neuronal transcription factor family PAS (Per-Arnt-Sim) domain has two distinct PAS domains that are protein 1 (NPAS1) and NPAS3. These are highly conserved structural sites of multi-ligand binding, responsible for physiological and potential pharmacological modulation in the brain (Zhou et al. 1997; E. W. Brunskill et al. 1999; Rutter et al. 2001; Dudley et al. 2003; Taylor and Zhulin 1999; Wu et al. 2016). Several studies have stated that gene NPAS3 is responsible for human evolution since it comprises of a huge number of human-specific, conserved, fast-evolving and non-coding elements (Kamm, Pisciottano, et al. 2013; Kamm, López-Leal, et al. 2013). It was found to be one of the 27 potent genes revealing repetitive double-stranded DNA break clusters into the neural stem cells. (Wei et al. 2016). The FGFR1 regulates the proliferation of hippocampal granule neurons in the mature hippocampal region of the brain in mouse where NPAS3 regulates the expression (Pieper et al. 2005). On the contrast, we understood from our study that NPAS1 prevents augmentation and plays a pivotal role in MAPK signaling in CGE and MGE progenitors, except not in the regulation of FGF receptor expression, but the process is not yet well known. Preclinical models illustrated the strong inclination towards NPAS3 and NPAS1 which had affected physiologic functioning of the neurons in the brain (Eric W. Brunskill et al. 2005; Erbel-Sieler et al. 2004; Yang et al. 2016). It also revealed the postnatal hippocampal neurogenesis by NPAS3 (Pieper et al. 2005) and significant impediment by NPAS1 (Pieper et al. 2005; Stanco et al. 2014). Many neuropsychiatric conditions are responsible due to several variations in Postnatal hippocampal neurogenesis (Nurnberger et al. 2014; Pieper et al. 2005; Kempermann, Krebs, and Fabel 2008). Studies have shown that mutations in NPAS1 in a set of individuals resulted in autism spectrum disorder (McKnight 2007). Whereas, NPAS3 anomalies were possibly implicated in several neuropsychiatric conditions, including neurological conditions like schizophrenia (Gonzalez-Penas et al.2015; Macintyre et al. 2010; B. S. Pickard et al. 2009; Ben S. Pickard et al. 2005; Kamnasaran et al. 2003; Benjamin S. Pickard et al. 2006), bipolar disorder (B. S. Pickard et al. 2009; Huang et al. 2010; Nurnberger et al. 2014), severe depression (Huang et al. 2010; Weber et al. 2011), attention deficit hyperactivity disorder (ADHD)(Weber et al. 2011) and hindrance in intellectual disability (Ben S. Pickard et al. 2005; Visser et al. 2010; Phelps et al. 2017).
In an experiment of the gene expression using microarray samples, it was seen that NPAS3 was overexpressed in HEK293 cells. HEK293 cell line had an unusual upregulation of NPAS3 which revealed that transcriptional targets differed according to two main factors; circadian rhythm context and Subsequent C-terminal deletion. Ultimately, the NPAS3 gene is highly overexpressed. After processing, The VGF and secretory peptides are known to have roles in neurogenesis and neuroplasticity which is related to learning, memory, cognition, depression, and schizophrenia (Sha et al. 2012). The Pathophysiology of NPAS can be noticeable as it perhaps triggered by an entirely new molecular pathway or maybe by a combinative effect of upsetting original pathways that are related to neuropsychiatric disorders.
Our present work focuses on visualization of gene-gene interactions of Neuronal Per Arnt Sim domain protein (NPAS) which is known to be associated with basic helix-loop-helix family, which is characterized as Npas1, Npas2, Npas3, and Npas4 (Ooe et al. 2004). This Neuronal family has been implicated to play a pivotal role in the epistasis of Autism Spectrum Disorder. Neuronal transcription factor family PAS (Per-Arnt-Sim) are mainly expressed in the brain Neuronal transcription factor family PAS (Per-Arnt-Sim) has two distinct PAS domains protein 1(NPAS1) and NPAS3. These genes have been identified to play a critical role in the development and maintenance of neurons in the brain (Jain 1999, Brunskill et al. 1999) Therefore, substantial studies were conducted using the UCSC browser as a filtering tool in which the top 24 high interacting genes with NPAS3 were identified. Among which only 8 genes showed maximum interlinkage with NPAS3. NPAS3 is known to have a role in Autism Spectrum Disorder and many other neurological diseases. The highly repeating interactions were screened. This study revealed that NPAS3 might not be the only one responsible but instead gene-gene interactions with CRKL might hold the key. The CRKL gene had the highest interacting gene among all the genes. Therefore, the CRKL gene emerged as the best possible candidate gene for ASD. Deletion or duplication of the CRKL gene can affect multiple genes leading to various birth defects such as Autism Spectrum Disorder, schizophrenia, heart, hearing, or autoimmune defects (Walz, Fonseca, and Lupski 2004). CRKL is a type of substrate created by BCR-ABL tyrosine kinase which is accountable for oncogenic transformations in fibroblast transformation. Computational tools had been used to predict a strong coherence for CRKL and its mutations, which later proposed that CRKL defects in the deleted region of 22q11. 2 of the human chromosome 22 hold the main key for several birth defects. It can change the development of the brain along with the behavior and cognitive functions leading to neuro-developmental disorders like autism spectrum disorder or schizophrenia, which might turn on a particular region of the gene and can cause further changes. The Action-Mechanism of the same is still not known. Homology search was performed using the human protein sequence of CRKL and it showed maximum homology with the horse (Figure 5).
Methodology
Top 24 interacting genes were efficiently screened by UCSC browser and several genes associated with such as- RPH3AL, SP1, TLX3, PATZ1, LEPREL4, PKN2, CFHR4, WWP2, MAP2K6, ADIPOR1, REST, SLC5A12, AMHR2, MED12, JARID2, ZNF787, RBBP7, PDLIM5, PINK1, CARD6, ARHGAP26, CCNE2, INHBE, and CRKL.
The workflow has been explained in Figure 1 and the gene-gene interactions have been visualized in Figure 2.
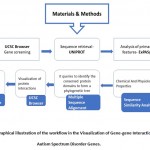 |
Figure 1: Graphical illustration of the workflow in the Visualization of Gene-gene Interactions in Autism Spectrum Disorder Genes. |
Results And Discussion
Through UCSC browser, these eight genes were known to be related to NPAS3 that are involved in neurodevelopment and other anomalies. They have been stated for its direct or indirect connection with Autism Spectrum Disorder- SYN1 (Paonessa et al. 2013b), TLX3 (Hernandez-Miranda et al. 2017) (Storm et al. 2009) (Qian 2001) , WWP2 (Kishimoto-Suga 2011), REST (Paonessa et al. 2013a), MED12 (Graham and Schwartz 2013), CRKL (Rapin and Katzman 1998)(Haller et al. 2017) , JARID2 (Ramos et al. 2012), PDLIM5 (Barh, Blum, and Madigan 2016)(Herrick et al. 2010) (Figure 2 and Table 1).
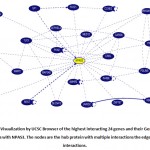 |
Figure 2: Visualization by UCSC Browser of the highest interacting 24 genes and their Gene-Gene Interactions with NPAS3. The nodes are the hub protein with multiple interactions the edges show the interactions. |
Table 1: The significance of highly interacting genes in neurological disorders.
| Sl.NO | Genes | Role in Neuropathology | References |
| 1 | SYN1 | Idiopathic epilepsy and autism | (Paonessa et al. 2013b) |
| 2 | TLX3 | Tlx3 mutations shown to affect the expansion of mostly non-overlapping neuronal populations. | (Hernandez-Miranda et al. 2017) (Storm et al. 2009) (Qian 2001) |
| 3 | WWP2 | WWP1/2performs various roles in synaptic function and development of neurons in the mammalian brain. Impairment of function can lead to neurological disorder | (Kishimoto-Suga 2011 |
| 4 | REST | It has as an important element connecting neuronal intrinsic homeostasis and re-establishing a physiological level of activity in the complete neuronal web. Dysfunction is published cause for neurological disorders. | Paonessa et al. 2013a) |
| 5 | MED12 | Member of the large Mediator complex which synchronizes signals involved in cell growth, development, and differentiation.
Extra neuronal gene silencing. |
(Graham and Schwartz 2013) |
| 6 | CRKL | CRKL has a crucial role in the growth of embryo, kidneys and testes development. Deletions or duplication cause multiple birth abnormalities. | (Rapin and Katzman 1998) (Haller et al. 2017) |
| 7 | JARID2 | Formation of neural tube
Necessary for normal heart function. |
(Ramos et al. 2012), |
| 8 | PDLIM5 | Bipolar disorder, schizophrenia and major depression PDLIM5 exists in the postsynaptic density, where it encourages reduced size of dendritic spine head and longer, filopodia-like morphology. | (Barh, Blum, and Madigan 2016) (Herrick et al. 2010) |
Table 2: The Prominent eight genes and their gene-gene interactions using UCSC browser.
| GENES | ASSOCIATED WITH OTHER GENES |
| SYN1 | SPTAN1, CALM2, MAPK4, REST, MAPK8, CALM1, MAPK7, CALM3, CA2, EPHB2, MAPK14, CYFIP1, MAPK12, MAPK3, MAPK1, MAPK11, MAPK13, MAPK6, BDNF, MAPK10, MAPK9, SLC6A4, MAPK15, NCF1 |
| TLX3 | TLE1, NUDT21, ETS1, SF3B5, HIST1H2BJ, CCNE2, SF3A3, PTPN14, RBBP6, CFHR4, WWP2, TLE2, MAP2K6, ING4, SYNE1, CRKL, AMHR2, ADIPOR1, MOCS3, TEKT2, JARID2, LEPREL4, PINK1, GTF2A1L |
| WWP2 | MIB2, HIST1H4A, HSPA6, HSPA8, TUBB4B, PTEN, MYL12A, DYNLL1, BYSL, CRKL, POU5F1, WDR5, ANP32B, SMARCC1, SLC5A12, SQRDL, DHX8, LEMD3, OPA1, PSPC1, ANP32A, USP34, RNF11, UBE4A |
| REST | KCNIP2, SP140, BARHL1, CAMTA1, ZNF444, BARHL2, MYT1, BDNF, EGR4, VSX1, MEIS3, NEUROG3, SF1, DAXX, GTF21, HIST1H4A, NR1H4, SIX6, GSX1, ONECUT2, MNX1, LHX4, SHOX2 |
| MED12 | POLR2J, RPAP1, POLR2B, SELP, ITGA2B, POLR2H, GTF2F1, POLR2C, LEP, POLR2A, INS, CTDP1, POLR2I, BDNF, EPHB2, POLR2D, POLR2E, TNFSF11, POLR2L, GTF2F2, GTF2B, POLR2G, POLR2F, POLR2F, POLR2K |
| CRKL | ITGAX, ITGA1, ITGAV, ITGAE, ITGA2, ITGA9, ITGA3, ITGA7, ITGAD, ITGA6, PIK3CG, JAK3, CBLC, PIK3CA, PIK3CD, JAK1, LAT, PIK3CB, ITGAM, RAP1B, ITGA8, ITGAL, MAP3K1, DBNL |
| JARID2 | ELL, AP1M1, WWP2, BCLAF1, CRKL, AGA, NPAS3, HIST1H3A, STK38, MORC3, EZH2, SUZ12, H3F3A, EED, HIST2H3A, TRIM35, HIST1H4A, HDAC2, MYL4, SETD1A, SOX17, RBBP7, RBBP4, QTRTD1 |
| PDLIM5 | HNRNPDL, ACYL, ANP32B, PKM, SUMO1, DIMT1, YBX1, RPS25, NAPG, RSU1, TUBB4B, SNAP23, DIAPH1, ANP32A, CAPZB, DDX47, SUPT5H, NAPA, ELAVL1, HSPA5, CRKL, NCBP1, DLG5, PDCD1O |
Interestingly notable, gene CRKL came out to be most interacting (4 out of 8 genes) among the highlighted genes (Table 2). The table marks the involvement of CRKL with TLX3, WWP2, JARID2, and PDLIM5. Hence, it clarifies the direct or indirect connection with neurological disorders as these eight genes are highly susceptible for neurological disorders especially with Autism Spectrum Disorder. Although its proper mechanism is not yet understood, the involvement of these genes cannot go unnoticed in neurological disorders.
 |
Table 3: Gene-Gene interaction between screened out genes and most frequently interacting genes with them (black in color). |
*Tables 2 and 3, show that CRKL is the gene, which interacts most with the highlighted genes. These results strongly point out to the involvement of direct or indirect connection with neurological disorders especially with Autism Spectrum Disorder. Further work on this gene is to see its physiological effects in Autistic patients.
The CRKL gene is situated in the typically deleted region of 22q11. 2. This region of human chromosome 22 holds the main possibility for many of the birth defects. It was observed that deletion of this region occurs in about 1 in 4,000 births, triggering the loss or duplication of up to 40 genes. Micro deletion or micro duplication in the chromosome can cause a number of developmental disorders that can differ in contrasting severity between the affected individuals. This region of the genes is not well understood, but the consequence of deleted or duplication in the set of these genes can create disruption in the development and function of the cognitive and behavioral functions along with heart, immune system, and craniofacial features. This condition is termed as 22q11.2 deletion syndrome or DiGeorge syndrome.
CRKL is expressed in a diversity of fetal tissues, including heart, spleen, thymus, brain, kidney as well as in liver, lung, and skeletal muscle which is relevant to DiGeorge syndrome. This gene is expressed moderately throughout developmental stages, including in the development of the genitourinary tract in mouse and human.
To decode the functioning of CRKL, scientists prepared model of genetically engineered mice with one copy of CRKL gene. Another group of mice received one copy from the mother and the other gene passed on by the father, lacking both copies of CRKL gene can be fatal for the developing embryos. This highlights the importance of CRKL in embryonic growth. The male mice lacked one copy of CRKL, had a failure of testicular descent into the scrotum which caused cryptorchidism in less-than-normal number of offsprings per litter, and when added with the aging process this subfertility grew into male infertility.
The research conclusions imply that patients with birth defects due to 22q11.2 abnormalities in the gene can give rise to other potential birth defects as seen in patients with DiGeorge syndrome which can affect future health. The influence of CRKL in the gene’s region 22q11.2 affect brain development and behavior and cognitive function, responsible for neurodevelopmental disorders such as bipolar disorder autism spectrum disorder, schizophrenia, heart, hearing or autoimmune defects depending on which particular gene in this region is affected (Haller et al. 2017).
Sequence Retrieval
In the sequence retrieval, we used CRKL (UNIPROT ID P46109) as a query sequence in further BLASTp searches. Precisely, it showed that BLASTp analysis against the non-redundant (nr) and PDB protein database results that the gene CRKL shares significant substantial sequence individuality and resemblance to mammalian proteins. [Table 4]
 |
Figure 3: Sequence of Human CRKL protein collected from Uniprot Database. |
Table 4: Analysis of Primary Features – Amino acid composition
| Amino acid | Symbol | No. | % |
| Ala | (A) | 20 | 6.6% |
| Arg | (R) | 18 | 5.9% |
| Asn | (N) | 16 | 5.3% |
| Asp | (D) | 16 | 5.3% |
| Cys | (C) | 2 | 0.7% |
| Gln | (Q) | 12 | 4.0% |
| Glu | (E) | 19 | 6.3% |
| Gly | (G) | 21 | 6.9% |
| His | (H) | 8 | 2.6% |
| Ile | (I) | 15 | 5.0% |
| Leu | (L) | 20 | 6.6% |
| Lys | (K) | 14 | 4.6% |
| Met | (M) | 6 | 2.0% |
| Phe | (F) | 11 | 3.6% |
| Pro | (P) | 30 | 9.9% |
| Ser | (5) | 24 | 7.9% |
| Thr | (1) | 15 | 5.0% |
| Trp | (W) | 4 | 1.3% |
| Tyr | (Y) | 12 | 4.0% |
| Val | (V) | 20 | 6.6% |
| Pyl | (0) | 0 | 0.0% |
| Sec | (U) | 0 | 0.0% |
| (B) | 0 | 0.0% | |
| (2) | 0 | 0.0% | |
| (X) | 0 | 0.0% |
Number of amino acids: 303
Molecular weight: 33776.99
Theoretical PI: 6.26
Total number of negatively charged residues (Asp + Glu): 35
Total number of positively charged residues (Arg + Lys): 32
Atomic Compositions
Carbon c 1504
Hydrogen H 2321
Nitrogen N 419
Oxygen O 453
Sulphur S 8
Total 4705
Formula: C1504H2321N419045358
Extinction coefficients: 4005
Abs 0.1% (=1 g/l) 1.184, assuming all pairs of Cys residues form from Cystines
Extinction coefficients: 39880
Abs 0.1% (=1 g/l) 1.181, assuming all pairs of Cys residues are reduced
The estimated half-life
Mammalian reticulocytes, in vitro: 30 hours
Yeast, in vivo: >20 hours
Escherichia coli, in vivo: >10 hours
Stability
The instability index (II) is computed to be 58.66 This classifies the protein as unstable.
Aliphatic Index: 70.79
Grand average of hydrophobicity: -0.582
 |
Figure 4: Physical Chemical feature of CRKL protein using Protparam Sequence Similarity Analyses. |
Expasy (protparam) were used to predict physiological and chemical features of CRKL (Table 4 and Figure 4). It states about the molecular weight, theoretical pI, atomic composition, amino acid composition, aliphatic index, grand average of hydropathicity, instability index, estimated half-life and extinction coefficient of query protein.
The BLASTp query analyses the sequence of CRKL for genomes of related mammals. The results showed that at least one protein shared a consequential sequence uniqueness and likeness as well as significant values, the final outcome was identified in 100% of the queried. While the number of CRKL genes per genome can differ according to species (Table 5)
Table 5: Homology of CRKL with other sequences using BLAST
| Selection | Description | Max Score | Total core | Query Cover | E Value | Per.Ident % | Accession |
| ü | Crk-Like Protein [Macau Mulatto]
|
631 | 631 | 100% | 0.0 | 100.00 | NP_0012444121 |
| PREDICTED: Crk-Like Protein [Chlorocebus Sabaeus] | 630 | 630 | 100% | 0.0 | 99.67 | P_007978127.1 | |
| ü | Chain A, Crk-Like Protein [Homo Sapiens] | 628 | 628 | 100% | 0.0 | 99.67 | 2 L OW_A |
| ü | Crk-Like Protein [Callithrix Jacchus] | 627 | 627 | 100% | 0.0 | 99.34 | X P_017833667 .1 |
| PREDICTED: Crk-Like Protein [Saimiri Boliviensis Boliviensis] | 626 | 626 | 100% | 0.0 | 99.01 | XP_003942698.1 | |
| ü | Crk-Like Protein [Tupaia Chinensis] | 625 | 625 | 100% | 0.0 | 98.35 | XP_006140254.2 |
| ü | Crk-Like Protein [Aotus Nancymaae] | 625 | 525 | 100% | 0.0 | 99.01 | XP_01 2321931.1 |
| CRIB Like Proto-Oncogene, Adaptor Protein [Molossus Molossus] | 624 | 624 | 100% | 0.0 | 98.68 | KAF6403223.1 | |
| ü | Crk-Like Protein [Sapajus Apella] | 624 | 624 | 100% | 0.0 | 98.68 | X P_032127229.1 |
| PREDICTED: Crk-Like Protein [Cebus Capucinus Imitator] | 624 | 624 | 100% | 0.0 | 98_35 | XP_017374120.1 | |
| PREDICTED: Crk-Like Protein [Miniopterus Natalensis] | 623 | 523 | 100% | 0.0 | 98_35 | P_01 6059814.1 | |
| ü | Crk-Like Protein [Tupaia Chinensis] | 622 | 622 | 100% | 0.0 | 98.35 | ELVV72575.1 |
| ü | Crk-Like Protein [Ailuropoda Melanolouca] | 621 | 621 | 100% | 0.0 | 98.02 | XP_002926331.1 |
| ü | Crk-Like Protein [Fukomys Damarensis] | 621 | 621 | 100% | 0.0 | 98.35 | P_01 0619076.1 |
| PREDICTED: Crk-Like Protein [Mustela Putorius Furo] | 621 | 621 | 100% | 0.0 | 98.02 | X P_004742614.1 | |
| Crk-Like Protein Isoform X1 [Castor Canadensis] | 621 | 621 | 100% | 0.0 | 98.02 | XP_02001 6330.1 | |
| ü | Crk-Like Protein [Desmodus Rotundus] | 620 | 620 | 100% | 0.0 | 98_02 | XP_024413072.1 |
| Crk-Like Protein Isoforni X1 [Eptesicus Fuscus] | 620 | 620 | 100% | 0.0 | 97.69 | X P_008140855.1 | |
| ü | Crk-Like Protein [Rhinolophus Ferrumequinum] | 620 | 620 | 100% | 0.0 | 97.36 | X P_032953699.1 |
| PREDICTED: Crk-Like Protein [Erinaceus Europaeus] | 620 | 620 | 100% | 0.0 | 98.02 | X P_007533835.1 | |
| ü | Crk-Like Protein [Suricato Suricatta] | 620 | 620 | 100% | 0.0 | 97.69 | X P_029778460. 1 |
Multiple Sequence Alignment
The primary sequence of CRKL was equated to a model of mammalian CRKLs by multiple sequence alignments and queries to recognize the conserved protein domains and to form a phylogenetic tree. From which we concluded that the human CRKL sequence is very similar to Horse CRKL sequence (Figure 5).
 |
Figure 5: Phylogenetic trees constructed from homologous sequence. |
Protein-Protein Interactions
The UCSC browser tool was used to identify protein interactions that CRKL interacts in vivo by Using default parameters of the browser. The result from the string clearly indicates that the CRKL gene is clearly connected to NPAS3 gene and along with its interacting genes which is directly or indirectly connected to the Autism Spectrum Disorder as its proper mechanism is not yet understood. CRKL has a strong influence in the fetal tissue which gives rise to a number of birth defects and chromosomal disorders. Autism can only be detected when the offspring is born which makes it difficult to detect the abnormalities. Notably CRKL can be a good target for detecting as it has been a hotspot for a variety of birth defects. Possible understanding and gene decoding can throw some light in the field of autism spectrum disorder. (Figure 6).
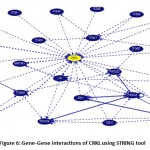 |
Figure 6: Gene-Gene interactions of CRKL using STRING tool. |
Conclusion
This study has brought focus on computational analysis to predict gene-gene interactions that were responsible for Autism Spectrum Disorder. The suspected genes seen after several filters showed that CRKL is the key gene responsible for the flaws in interaction. During the analysis of the interactions between the genes CRKL along with other highlighted genes which are responsible for autism spectrum disorder and other neurological diseases. CRKL interacted most with 4 out of 8 highlighted genes like TLX3, WWP2, JARID2, and PDLIM5. This interaction can play a pivotal role directly or indirectly in the disorder. CRKL is a substrate formed by BCR-ABL tyrosine kinase which is responsible for fibroblast transformation by BCR-ABL, that might be oncogenic in behavior. It encodes for a protein kinase that consists of two main domains SH2 and SH3 (src homology). There are mainly two signaling pathways, the RAS and JUN kinase. They are known to be triggered, consequently resulting in the formation as changes in fibroblasts in a RAS-dependent form. Evidence indicating RAS signaling in autism attain from the effective studies of particular genes and from comparisons between autism and condition due to hyperactivation of the RAS pathway — the RASopathies. Gary Landreth and his fellow workers observed that the collection of phenotypes linked with 16p11.2 deletion — obstruction in the central nervous system, heart and brain, along with facial dysmorphology — is also seen in the RASopathies, comprising DiGeorge syndrome that is caused by the 22q11.2 chromosomal region 4 deletions. Various Bioinformatics tools were used that predict strong coherence for CRKL and its mutations it suggested that CRKL defects in the deleted region of 22q11. 2 of the human chromosome 22 hold the main potential for several of the birth defects. It can hamper the development of brain along with behavior and cognitive function leading to neurodevelopmental disorders like autism spectrum disorder or schizophrenia, depending on which particular region of the gene is affected. After performing homology study using human protein sequence of CRKL it surprisingly showed maximum homology with the horse genome. The role of CRKL during prenatal stages is vital for the growth and development of the embryo. Therefore, identifying any mutations during these phases can be a potential therapeutic intervention in Autism Spectrum Disorder and other neurological disorders. CRKL gene can be a potential biomarker for several birth defects. Future studies are required to characterize and understand the mechanism of action of the CRKl gene and the biological roles via wet lab and dry lab studies for deeper understanding.
References
- Abrahams, Brett S., and Daniel H. Geschwind. 2008. “Advances in Autism Genetics: On the Threshold of a New Neurobiology.” Nature Reviews. Genetics 9 (5): 341–55.
- Barh, Debmalya, Kenneth Blum, and Margaret A. Madigan. 2016. OMICS: Biomedical Perspectives and Applications. CRC Press.
- Beaudet, Arthur L. 2007. “Autism: Highly Heritable but Not Inherited.” Nature Medicine. https://doi.org/1038/nm0507-534.
- Bonnet-Brilhault, F. 2017. “Autism: An early neurodevelopmental disorder.” Archives of pediatrics 24 (4): 384–90.
- Brunskill, E. W., D. P. Witte, A. B. Shreiner, and S. S. Potter. 1999. “Characterization of npas3, a Novel Basic Helix-Loop-Helix PAS Gene Expressed in the Developing Mouse Nervous System.” Mechanisms of Development 88 (2): 237–41.
- Brunskill, Eric W., David P. Witte, Andrew B. Shreiner, and S. Steven Potter. 1999. “Characterization of Npas3 , a Novel Basic Helix-Loop-Helix PAS Gene Expressed in the Developing Mouse Nervous System.” Mechanisms of Development. https://doi.org/1016/s0925-4773(99)00182-3.
- Brunskill, Eric W., Lisa A. Ehrman, Michael T. Williams, Justin Klanke, Daniel Hammer, Tori L. Schaefer, Renu Sah, Gerald W. Dorn 2nd, S. Steven Potter, and Charles V. Vorhees. 2005. “Abnormal Neurodevelopment, Neurosignaling and Behaviour in Npas3-Deficient Mice.” The European Journal of Neuroscience 22 (6): 1265–76.
- Consortium, The International Schizophrenia, and The International Schizophrenia Consortium. 2009. “Common Polygenic Variation Contributes to Risk of Schizophrenia and Bipolar Disorder.” Nature. https://doi.org/1038/nature08185.
- Drgon, Tomas, Ivan Montoya, Catherine Johnson, Qing-Rong Liu, Donna Walther, Dean Hamer, and George R. Uhl. 2009. “Genome-Wide Association for Nicotine Dependence and Smoking Cessation Success in NIH Research Volunteers.” Molecular Medicine. https://doi.org/2119/molmed.2008.00096.
- Dudley, Carol A., Claudia Erbel-Sieler, Sandi Jo Estill, Martin Reick, Paul Franken, Sinae Pitts, and Steven L. McKnight. 2003. “Altered Patterns of Sleep and Behavioral Adaptability in NPAS2-Deficient Mice.” Science 301 (5631): 379–83.
- Erbel-Sieler, Claudia, Carol Dudley, Yudong Zhou, Xinle Wu, Sandi Jo Estill, Tina Han, Ramon Diaz-Arrastia, Eric W. Brunskill, S. Steven Potter, and Steven L. McKnight. 2004. “Behavioral and Regulatory Abnormalities in Mice Deficient in the NPAS1 and NPAS3 Transcription Factors.” Proceedings of the National Academy of Sciences of the United States of America 101 (37): 13648–53.
- Fombonne, Eric. 2009. “Epidemiology of Pervasive Developmental Disorders.” Pediatric Research. https://doi.org/1203/pdr.0b013e31819e7203.
- Gai, X., H. M. Xie, J. C. Perin, N. Takahashi, K. Murphy, A. S. Wenocur, M. D’arcy, et al. 2012. “Rare Structural Variation of Synapse and Neurotransmission Genes in Autism.” Molecular Psychiatry 17 (4): 402–11.
- González-Peñas, Javier, Manuel Arrojo, Eduardo Paz, Julio Brenlla, Mario Páramo, and Javier Costas. 2015. “Cumulative Role of Rare and Common Putative Functional Genetic Variants at NPAS3 in Schizophrenia Susceptibility.” American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics: The Official Publication of the International Society of Psychiatric Genetics 168 (7): 528–35.
- Graham, John M., Jr, and Charles E. Schwartz. 2013. “MED12 Related Disorders.” American Journal of Medical Genetics. Part A 161A (11): 2734–40.
- Hagerman, Randi, Susan Rivera, and Paul Hagerman. 2008. “The Fragile X Family of Disorders: A Model for Autism and Targeted Treatments.” Current Pediatric Reviews. https://doi.org/2174/157339608783565770.
- Haller, Meade, Qianxing Mo, Akira Imamoto, and Dolores J. Lamb. 2017. “Murine Model Indicates 22q11.2 Signaling Adaptor CRKL Is a Dosage-Sensitive Regulator of Genitourinary Development.” Proceedings of the National Academy of Sciences. https://doi.org/1073/pnas.1619523114.
- Haller, Meade, Qianxing Mo, Akira Imamoto, and Dolores J. Lamb. 2017. “Murine Model Indicates 22q11.2 Signaling Adaptor CRKL Is a Dosage-Sensitive Regulator of Genitourinary Development.” Proceedings of the National Academy of Sciences. https://doi.org/1073/pnas.1619523114.
- Hernandez-Miranda, Luis Rodrigo, Pierre-Louis Ruffault, Julien C. Bouvier, Andrew J. Murray, Marie-Pierre Morin-Surun, Niccolò Zampieri, Justyna B. Cholewa-Waclaw, et al. 2017. “Genetic Identification of a Hindbrain Nucleus Essential for Innate Vocalization.” Proceedings of the National Academy of Sciences of the United States of America 114 (30): 8095–8100.
- Herrick, Scott, Danielle M. Evers, Ji-Yun Lee, Noriko Udagawa, and Daniel T. S. Pak. 2010. “Postsynaptic PDLIM5/Enigma Homolog Binds SPAR and Causes Dendritic Spine Shrinkage.” Molecular and Cellular Neurosciences 43 (2): 188–200.
- Huang, Jie, Roy H. Perlis, Phil H. Lee, A. John Rush, Maurizio Fava, Gary S. Sachs, Jeffrey Lieberman, et al. 2010. “Cross-Disorder Genomewide Analysis of Schizophrenia, Bipolar Disorder, and Depression.” The American Journal of Psychiatry 167 (10): 1254–63.
- Jain, Sanjay. 1999. Characterization of BHLH-PAS Superfamily of Transcription Factors.
- Kamm, Gretel B., Francisco Pisciottano, Rafi Kliger, and Lucía F. Franchini. 2013. “The Developmental Brain Gene NPAS3 Contains the Largest Number of Accelerated Regulatory Sequences in the Human Genome.” Molecular Biology and Evolution 30 (5): 1088–1102.
- Kamm, Gretel B., Rodrigo López-Leal, Juan R. Lorenzo, and Lucía F. Franchini. 2013. “A Fast-Evolving Human NPAS3 Enhancer Gained Reporter Expression in the Developing Forebrain of Transgenic Mice.” Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 368 (1632): 20130019.
- Kamnasaran, D., W. J. Muir, M. A. Ferguson-Smith, and D. W. Cox. 2003. “Disruption of the Neuronal PAS3 Gene in a Family Affected with Schizophrenia.” Journal of Medical Genetics 40 (5): 325–32.
- Kaufmann, Walter E., Ranon Cortell, Alice S. M. Kau, Irena Bukelis, Elaine Tierney, Robert M. Gray, Christiane Cox, George T. Capone, and Pia Stanard. 2004. “Autism Spectrum Disorder in Fragile X Syndrome: Communication, Social Interaction, and Specific Behaviors.” American Journal of Medical Genetics. Part A 129A (3): 225–34.
- Kempermann, Gerd, Julia Krebs, and Klaus Fabel. 2008. “The Contribution of Failing Adult Hippocampal Neurogenesis to Psychiatric Disorders.” Current Opinion in Psychiatry 21 (3): 290–95.
- Kishimoto-Suga, Mika. 2011. “The Role of the HECT-Type Ubiquitin Ligases WWP1 and WWP2 in Nerve Cell Development and Function.” Doctor of Philosophy (PhD), Georg August University Göttingen. https://d-nb.info/1043999159/34.
- Macintyre, Georgina, Tyler Alford, Lan Xiong, Guy A. Rouleau, Philip G. Tibbo, and Diane W. Cox. 2010. “Association of NPAS3 Exonic Variation with Schizophrenia.” Schizophrenia Research 120 (1-3): 143–49.
- McKnight, Steven. 2007. “Relevance of NPAS1/3 Balance to Autism and Schizophrenia.” SFARI, October. https://www.sfari.org/funded-project/relevance-of-npas13-balance-to-autism-and-schizophrenia/.
- Mitra, Ileena, Alinoë Lavillaureix, Erika Yeh, Michela Traglia, Kathryn Tsang, Carrie E. Bearden, Katherine A. Rauen, and Lauren A. Weiss. 2017. “Reverse Pathway Genetic Approach Identifies Epistasis in Autism Spectrum Disorders.” PLoS Genetics 13 (1): e1006516.
- Nurnberger, John I., Daniel L. Koller, Jeesun Jung, Howard J. Edenberg, Tatiana Foroud, Ilaria Guella, Marquis P. Vawter, and John R. Kelsoe. 2014. “Identification of Pathways for Bipolar Disorder: A Metaanalysis.” JAMA Psychiatry 71 (6): 657.
- Ooe, Norihisa, Koichi Saito, Nobuyoshi Mikami, Iwao Nakatuka, and Hideo Kaneko. 2004. “Identification of a Novel Basic Helix-Loop-Helix-PAS Factor, NXF, Reveals a Sim2 Competitive, Positive Regulatory Role in Dendritic-Cytoskeleton Modulator Drebrin Gene Expression.” Molecular and Cellular Biology. https://doi.org/1128/mcb.24.2.608-616.2004.
- Paonessa, Francesco, Shahrzad Latifi, Helena Scarongella, Fabrizia Cesca, and Fabio Benfenati. 2013. “Specificity Protein 1 (Sp1)-Dependent Activation of the Synapsin I Gene (SYN1) Is Modulated by RE1-Silencing Transcription Factor (REST) and 5′-Cytosine-Phosphoguanine (CpG) Methylation.” Journal of Biological Chemistry. https://doi.org/1074/jbc.m112.399782.
- Paonessa, Francesco, Shahrzad Latifi, Helena Scarongella, Fabrizia Cesca, and Fabio Benfenati. 2013a. “Specificity Protein 1 (Sp1)-Dependent Activation of the Synapsin I Gene (SYN1) Is Modulated by RE1-Silencing Transcription Factor (REST) and 5′-Cytosine-Phosphoguanine (CpG) Methylation.” Journal of Biological Chemistry. https://doi.org/1074/jbc.m112.399782.
- Phelps, Randall, Anne Tsai, Arlene Hagen, Joseph Pinter, Raegan Smith, and Martin T. Stein. 2017. “The Curse of the Dolphins: Cognitive Decline and Psychosis.” Journal of Developmental and Behavioral Pediatrics: JDBP 38 Suppl 1: S16–18.
- Pickard, B. S., A. Christoforou, P. A. Thomson, A. Fawkes, K. L. Evans, S. W. Morris, D. J. Porteous, D. H. Blackwood, and W. J. Muir. 2009. “Interacting Haplotypes at the NPAS3 Locus Alter Risk of Schizophrenia and Bipolar Disorder.” Molecular Psychiatry 14 (9): 874–84.
- Pickard, Ben S., M. P. Malloy, D. J. Porteous, D. H. R. Blackwood, and W. J. Muir. 2005. “Disruption of a Brain Transcription Factor, NPAS3, Is Associated with Schizophrenia and Learning Disability.” American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. https://doi.org/1002/ajmg.b.30204.
- Pickard, Benjamin S., Andrew A. Pieper, David J. Porteous, Douglas H. Blackwood, and Walter J. Muir. 2006. “TheNPAS3gene—emerging Evidence for a Role in Psychiatric Illness.” Annals of Medicine 38 (6): 439–48.
- Pieper, Andrew A., Xinle Wu, Tina W. Han, Sandi Jo Estill, Quyen Dang, Leeju C. Wu, Sarah Reece-Fincanon, et al. 2005. “The Neuronal PAS Domain Protein 3 Transcription Factor Controls FGF-Mediated Adult Hippocampal Neurogenesis in Mice.” Proceedings of the National Academy of Sciences of the United States of America 102 (39): 14052–57.
- Qian, Y. 2001. “Formation of Brainstem (nor)adrenergic Centers and First-Order Relay Visceral Sensory Neurons Is Dependent on Homeodomain Protein Rnx/Tlx3.” Genes & Development. https://doi.org/1101/gad.921501.
- Ramos, Paula S., Satria Sajuthi, Carl D. Langefeld, and Stephen J. Walker. 2012. “Immune Function Genes CD99L2, JARID2 and TPO Show Association with Autism Spectrum Disorder.” Molecular Autism 3 (1): 4.
- Rapin, Isabelle, and Robert Katzman. 1998. “Neurobiology of Autism.” Annals of Neurology. https://doi.org/1002/ana.410430106.
- Rogers, S. J., D. E. Wehner, and R. Hagerman. 2001. “The Behavioral Phenotype in Fragile X: Symptoms of Autism in Very Young Children with Fragile X Syndrome, Idiopathic Autism, and Other Developmental Disorders.” Journal of Developmental and Behavioral Pediatrics: JDBP 22 (6): 409–17.
- Rutter, J., M. Reick, L. C. Wu, and S. L. McKnight. 2001. “Regulation of Clock and NPAS2 DNA Binding by the Redox State of NAD Cofactors.” Science 293 (5529): 510–14.
- Samuels, Ivy S., Sulagna C. Saitta, and Gary E. Landreth. 2009. “MAP’ing CNS Development and Cognition: An ERKsome Process.” Neuron. https://doi.org/1016/j.neuron.2009.01.001.
- Sha, L., L. MacIntyre, J. A. Machell, M. P. Kelly, D. J. Porteous, N. J. Brandon, W. J. Muir, et al. 2012. “Transcriptional Regulation of Neurodevelopmental and Metabolic Pathways by NPAS3.” Molecular Psychiatry 17 (3): 267–79.
- Shihab, Ammar I., Faten A. Dawood, and Ali H. Kashmar. 2020. “Data Analysis and Classification of Autism Spectrum Disorder Using Principal Component Analysis.” Advances in Bioinformatics 2020 (January): 3407907.
- Stanco, Amelia, Ramón Pla, Daniel Vogt, Yiran Chen, Shyamali Mandal, Jamie Walker, Robert F. Hunt, et al. 2014. “NPAS1 Represses the Generation of Specific Subtypes of Cortical Interneurons.” Neuron 84 (5): 940–53.
- Storm, R., J. Cholewa-Waclaw, K. Reuter, D. Brohl, M. Sieber, M. Treier, T. Muller, and C. Birchmeier. 2009. “The bHLH Transcription Factor Olig3 Marks the Dorsal Neuroepithelium of the Hindbrain and Is Essential for the Development of Brainstem Nuclei.” Development. https://doi.org/1242/dev.027193.
- Sykes, Nuala H., and Janine A. Lamb. 2007. “Autism: The Quest for the Genes.” Expert Reviews in Molecular Medicine 9 (24): 1–15.
- Taylor, B. L., and I. B. Zhulin. 1999. “PAS Domains: Internal Sensors of Oxygen, Redox Potential, and Light.” Microbiology and Molecular Biology Reviews: MMBR 63 (2): 479–506.
- Visser, Remco, Antoinet Gijsbers, Claudia Ruivenkamp, Marcel Karperien, H. Maarten Reeser, Martijn H. Breuning, Sarina G. Kant, and Jan M. Wit. 2010. “Genome-Wide SNP Array Analysis in Patients with Features of Sotos Syndrome.” Hormone Research in Paediatrics 73 (4): 265–74.
- Walz, Katherina, Patricia Fonseca, and James R. Lupski. 2004. “Animal Models for Human Contiguous Gene Syndromes and Other Genomic Disorders.” Genetics and Molecular Biology. https://doi.org/1590/s1415-47572004000300001.
- Weber, Heike, Sarah Kittel-Schneider, Alexandra Gessner, Katharina Domschke, Maria Neuner, Christian P. Jacob, Henriette N. Buttenschon, et al. 2011. “Cross-Disorder Analysis of Bipolar Risk Genes: Further Evidence of DGKH as a Risk Gene for Bipolar Disorder, but Also Unipolar Depression and Adult ADHD.” Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology 36 (10): 2076–85.
- Wei, Pei-Chi, Amelia N. Chang, Jennifer Kao, Zhou Du, Robin M. Meyers, Frederick W. Alt, and Bjoern Schwer. 2016. “Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells.” Cell 164 (4): 644–55.
- Wu, Dalei, Xiaoyu Su, Nalini Potluri, Youngchang Kim, and Fraydoon Rastinejad. 2016. “NPAS1-ARNT and NPAS3-ARNT Crystal Structures Implicate the bHLH-PAS Family as Multi-Ligand Binding Transcription Factors.” eLife 5 (October). https://doi.org/7554/eLife.18790.
- Yang, Dongxue, Wenbo Zhang, Arshad Padhiar, Yao Yue, Yonghui Shi, Tiezheng Zheng, Kaspar Davis, et al. 2016. “NPAS3 Regulates Transcription and Expression of VGF: Implications for Neurogenesis and Psychiatric Disorders.” Frontiers in Molecular Neuroscience 9 (November): 109.
- Zhou, Y. D., M. Barnard, H. Tian, X. Li, H. Z. Ring, U. Francke, J. Shelton, J. Richardson, D. W. Russell, and S. L. McKnight. 1997. “Molecular Characterization of Two Mammalian bHLH-PAS Domain Proteins Selectively Expressed in the Central Nervous System.” Proceedings of the National Academy of Sciences of the United States of America 94 (2): 713–18.
- Zuk, O., E. Hechter, S. R. Sunyaev, and E. S. Lander. 2012. “The Mystery of Missing Heritability: Genetic Interactions Create Phantom Heritability.” Proceedings of the National Academy of Sciences. https://doi.org/10.1073/pnas.1119675109.

This work is licensed under a Creative Commons Attribution 4.0 International License.



