

Manuscript accepted on : 13-Nov-2021
Published online on: 14-12-2021
Plagiarism Check: Yes
Reviewed by: Dr. Rania I.M. Almoselhy
Second Review by: Dr. Revathi Shenoy
Final Approval by: Dr. Dai-Viet No, Dr. Fernando José Cebola Lidon
Rufiat Nahar , Alam MD Noor A , Islam MD Alrazi and Shinsaku Maruta*
Division of Bioinformatics, Graduate School of Engineering, Soka University, Hachioji, Tokyo 192-8577, Japan
Corresponding Author E-mail: maruta@soka.ac.jp
DOI : http://dx.doi.org/10.13005/bbra/2949

ABSTRACT:
Ras is a small G protein known as a central regulator of cellular signal transduction that induces processes, such as cell division, transcription. The hypervariable region (HVR) is one of the functional parts of this G protein, which induces multimerization and interaction between Ras and the plasma membrane. We introduced two highly different in polarity photochromic SH group-reactive azobenzene derivatives, N-4-phenyl-azophenyl maleimide (PAM) and 4-chloroacetoamido-4-sulfo-azobenzene (CASAB), into three cysteine residues in HVR to control Ras GTPase using light. PAM stoichiometrically reacted with the SH group of cysteine residues and induced multimerization. The mutants modified with PAM exhibited reversible changes in GTPase activity accelerated by the guanine nucleotide exchange factor and GTPase activating protein and multimerization accompanied by cis- and trans-photoisomerization upon ultraviolet and visible light irradiation. CASAB was incorporated into two of the three cysteine residues in HVR but did not induce multimerization. The H-Ras GTPase modified with CASAB was photo controlled more effectively than PAM-H-Ras. In this study, we revealed that the incorporation of azobenzene derivatives into the functional site of HVR enables photo reversible control of Ras function. Our findings may contribute to the development of a method to control functional biomolecules with physiologically important roles.
KEYWORDS: Azobenzene Derivatives; Chemical Modification; Photochromic Molecule; Ras; Small Gtpase
Download this article as:| Copy the following to cite this article: Nahar R, Noor A. A. M. D, Alrazi I. M. D, Maruta S. Photocontrol of GTPase Cycle and Multimerization of the Small G-Protein H-Ras using Photochromic Azobenzene Derivatives. Biosci Biotech Res Asia 2021;18(4). |
| Copy the following to cite this URL: Nahar R, Noor A. A. M. D, Alrazi I. M. D, Maruta S. Photocontrol of GTPase Cycle and Multimerization of the Small G-Protein H-Ras using Photochromic Azobenzene Derivatives. Biosci Biotech Res Asia 2021;18(4). Available from: https://bit.ly/3GGBzAl |
Introduction
The small GTPase Ras acts as a binary switch in the signal transduction pathways to control cell growth, differentiation, and proliferation by cycling between the GDP-bound “switched off state” and the GTP-bound “switched on state” 1, 2. Two factors regulate the active and inactive states of Ras. GTPase-activating protein (GAP) induces the hydrolysis of GTP to GDP, inactivating Ras. On the other hand, the guanine nucleotide exchange factor (GEF) replaces GDP with GTP and switches Ras on. Switching mechanisms utilizing conformational changes in nucleotide-binding motifs have been well studied at the molecular level 3. Interestingly, recent structural studies have indicated that G proteins contain a conserved nucleotide-binding motif with ATP-driven motors: kinesin, and myosin 4. These nucleotide-binding proteins might have evolved from a common nucleotide-binding ancestral protein, as they share a common catalytic core region, including the P-loop, Switch I, and Switch II, and molecular mechanisms utilizing a nucleotide hydrolysis cycle. Previously, we demonstrated that incorporating artificial regulatory nanodevices, such as photochromic molecules, into the kinesin functional site enables photo reversible regulation of ATPase activity 5-7. Subsequently, we incorporated azobenzene derivatives into the functional sites of the motor domain and succeeded in photo controlling GTPase activity 8. H-Ras has a specific unique functional domain in the hypervariable region (HVR) located at the C-terminus, which plays a physiologically important role. It is known that cysteine residues in HVR are modified by farnesylation and palmitoylation. Lipidation enables H-Ras to bind to the membrane and form clusters, resulting in intracellular signal transduction. It is known that the location of lipidated HVR in the globular domain is different from that of the unlipidated H-Ras 9, 10. Therefore, conformational changes in the HVR determine the physiological performance of Ras. We have recently demonstrated that chemical modification of the cysteine residues in the HVR of Ras with SH group-reactive caged compounds instead of lipidation induces multimerization of Ras, which may mimic the physical cluster structure 11. The multimer returned to the monomer by eliminating the caged group from the HVR by light irradiation. The results suggested that incorporating a specific group into the HVR lipidation site may induce artificial physiological structural changes related to Ras function. However, the caged compound exhibits an irreversible photoelimination reaction 12. Therefore, the caged components do not work as reversible photos witches. In the present study, we utilized an azobenzene derivative as a photo-reversible nano-switching device and incorporated it into the HVR to control Ras function.
Material and Methods
Toyobo (Tokyo) restriction enzymes and related enzymes were used. The designed cDNA was synthesized using Integrated DNA Technologies (Japan). Affinity chromatography using Co2+-NTA agarose (Clontech, CA, USA) was used to purify the recombinant Ras and related proteins. N-(4-phenylazophenyl) maleimide (PAM) was purchased from Sigma-Aldrich (St. Louis, MO, USA). BIOMOL GREEN was purchased from Enzo (Tokyo, Japan).
Expression and purification of the Ras mutant
Human H-Ras WT (1-189) cDNA plasmids offered by Dr. Sako (RIKEN) were amplified using PCR and incorporated into the pET42c vector using the general ligation procedure. We constructed a plasmid of the mutant H-Ras, where cysteine 118 was substituted by serine (C118S) to delete the reactive cysteine residue in the globular domain of Ras. Ras expression plasmids were transformed into Escherichia coli Rosetta2 (Invitrogen, Carlsbad, CA, USA). The Ras protein was purified according to established methods using a Co2+-NTA column. Purified Ras in the stock solution buffer [150 mM NaCl, 1 mM MgCl2, 30 mM Tris-HCl (pH 7.5), and 0.5 mM DTT] was stored in a deep freezer.
Expression and purification of GEF and NF1
GEF and NF1 (GAP GRD domain) cDNA was designed with a multi-cloning site for three different vectors. The two vectors, pET-21a and pET-15b, were chosen based on their availability in the laboratory. The designed cDNA was then synthesized by Integrated DNA Technologies (IDT, Japan). The NF1 and SOS cDNA synthesized with a multi-cloning site was treated with the chosen restriction enzymes and ligated into the pET-21a and pET-15b vectors, respectively. NF1 and GEF expression plasmids were transformed into Escherichia coli Rosetta2 (DE3) pLysS (Invitrogen, CA, USA). GEF and NF1 were purified using a Co2+-NTA column. Purified NF1 was stored in a buffer solution containing 150 mM NaCl, 1 mM MgCl2, 30 mM Tris-HCl (pH 7.5) and 0.5 mM DTT in a deep freezer (-80 °C). Purified GEF was stored in a buffer containing 150 mM NaCl, 1 mM MgCl2, 30 mM Tris-HCl (pH 7.5), 0.5 mM DTT, and 0.1% CHAPS in a deep freezer (-80 °C) until further use.
Synthesis of 4-chloroacetoamido-4-sulfo-azobenzene (CASAB)
CASAB synthesis was performed in a single-step reaction between 4-aminoazobenzene-4′-sulfonic acid sodium salt (167.06 μmol) and chloroacetic anhydride (417.65 μmol). The ratio of the reaction was 1:2.5, and it was left to react for 30 min in 1 ml dry N, N-dimethylformamide (DMF) at 25 °C and stirred with a magnetic stirrer. The expected product was purified with a large TLC (PLC silica gel 60, MERCK) with 20% methanol and 80% chloroform as a developing solvent. CASAB showed an Rf value of 0.26. The synthesis of CASAB was confirmed by fast atom bombardment mass spectroscopy. The products showed a molecular ion at m/z 352+1, corresponding to the molecular mass of 353.781 of the CASAB formula.
Photoirradiation for isomerization of Ras modified with PAM and CASAB
The isomerization of PAM and CASAB was performed at 0 °C using UV light irradiation generated with a Blak-Ray lamp (16 W) (UVP Inc., San Gabriel, USA) at 366 nm for induction of the cis state and by visible light irradiation using a room fluorescent lamp (27 W) to induce the trans state.
Modification of the H-Ras HVR domain using PAM and CASAB
The modification of the HVR domain with PAM was performed as follows: H-Ras (5 mM) reacted with PAM (18 mM) in the reaction buffer (120 mM NaCl, 30 mM Tris-HCl pH 7.5) with 3% DMF at 25 °C for 10 min. The reaction was stopped by the addition of DTT to a final concentration of 2 mM. Subsequently, the unreacted reagents were removed using a gel filtration column (10 DG; Bio-Rad, Hercules, CA, USA) pre-packed with a buffer containing 30 mM Tris-HCl (pH 7.5) and 120 mM NaCl. The stoichiometric incorporation of PAM was estimated using the PAM extinction coefficient (10,620 M-1 cm-1 at 350 nm). The HVR domain was also modified by H-Ras (5 mM) and CASAB (40 mM) reaction in the reaction buffer (120 mM NaCl, 30 mM Tris-HCl pH 8.0) with 3% DMF for 12 h at 25 °C in the dark. The reaction was stopped by the addition of DTT to a final concentration of 2 mM. The modified H-Ras was separated from the unreacted reagents using a gel filtration column (10 DG; Bio-Rad, Hercules, CA, USA) equilibrated with 30 mM Tris-HCl buffer (pH 8.0) and 120 mM NaCl. Stoichiometric incorporation of CASAB was estimated according to the same method used for PAM.
Measurement of GTPase activity
GTPase activity was determined using a previously established method. H-Ras (2 μM) in GTPase activity assay buffer (30 mM Tris-HCl pH 7.5, 60 mM NaCl, 2 mM MgCl2) was pre-incubated for 5 min in the presence of 2 μM GAP and GEF. Subsequently, 1 mM GTP was added to initiate the GTPase assay at 25 °C for 30 min, and the reaction was stopped by the addition of 10 % trichloroacetic acid. After centrifugation for 5 min at 15000 rpm at 4 °C, the supernatant was mixed with BioMol Green Reagent and incubated at 25 °C for 30 min to quantitatively determine the amount of Pi generated by GTP hydrolysis. The cis isomer of Ras modified with PAM or CASAB was induced by UV light irradiation (Blak-Ray lamp, 366 nm, 16 W, UVP Inc., San Gabriel, CA, USA) for 3 min. Ras modified with PAM or CASAB was irradiated with visible light (Fluorescent lamp 27 W, FML27EX-N, Mitsubishi Inc., JP) for 10 min to convert to the trans isomer. The GTPase activity of Ras modified with PAM or CASAB was measured at 25 °C in GTPase assay buffer.
Size-exclusion column chromatography coupled with high-performance liquid chromatography (SEC-HPLC)
SEC-HPLC was performed on a TSKgel G3000PWXL column (TOSOH, Tokyo, Japan, φ7.5 mm x 30 cm) or a TSKgel SuperSW3000 column (TOSOH, Tokyo, Japan, v4.6 mm x 30 cm). The 0.5 mg/ml samples were eluted with size-exclusion buffer, 30 mM Tris-HCl pH 7.5, and 120 mM NaCl at a flow rate of 1.0 or 0.4 ml/min while monitoring the absorbance at 280 nm.
Results
Modification of cysteine residues in HVR with azobenzene derivatives
We utilized two thiol-reactive azobenzene derivatives, PAM and CASAB to incorporate cysteine residues as lipidation sites in HVR in order to photocontrol the H-Ras function. As shown in Figure 1, PAM and CASAB exhibited cis and trans isomerization under UV and visible light irradiation, respectively. PAM is relatively hydrophobic and is expected to have an effect similar to lipidation. On the other hand, CASAB contains a highly negatively charged sulfo group. To determine the optimal conditions for labeling the cysteine residues in HVR with azobenzene derivatives, time course and concentration dependence reactions were examined. In this study, we prepared an H-Ras mutant, C118S, where the C118 cysteine residue on the globular domain surface was replaced by serine. Therefore, the H-Ras mutant contained three reactive cysteine residues in HVR. The incorporation of PAM into H-Ras was very quickly saturated with modification time >1 min (data not shown), consistent with kinesin and calmodulin interaction with PAM 13, 5. The PAM concentration-dependent experiment indicated that the modification of Ras with PAM was saturated at approximately four times the amount of Ras, as shown in Figure 3A. The three cysteine residues in the HVR of H-Ras mutants were almost stoichiometrically modified by PAM (Cys residues in HVR:PAM = 3.0:3.2). In contrast, the reaction with CASAB took more than 10 h to saturate the HVR. Interestingly, the incorporation of CASAB into C118S showed saturation at a ratio of Cys residues in HVR:CASAB = 3:2. These results suggest that two among the three cysteine residues in HVR were specifically modified. As shown in Figure 2, the cysteine residues in the HVR were located very close to each other. Therefore, a possible explanation for the insufficient stoichiometric incorporation is the strong intermolecular negative charge repulsion between the sulfonate groups in the CASAB molecules.
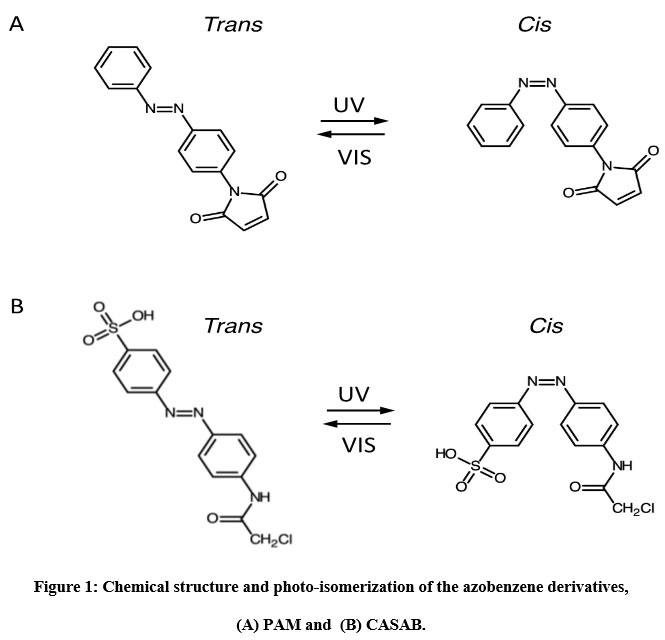 |
Figure 1: Chemical structure and photo-isomerization of the azobenzene derivatives, (A) PAM and (B) CASAB. |
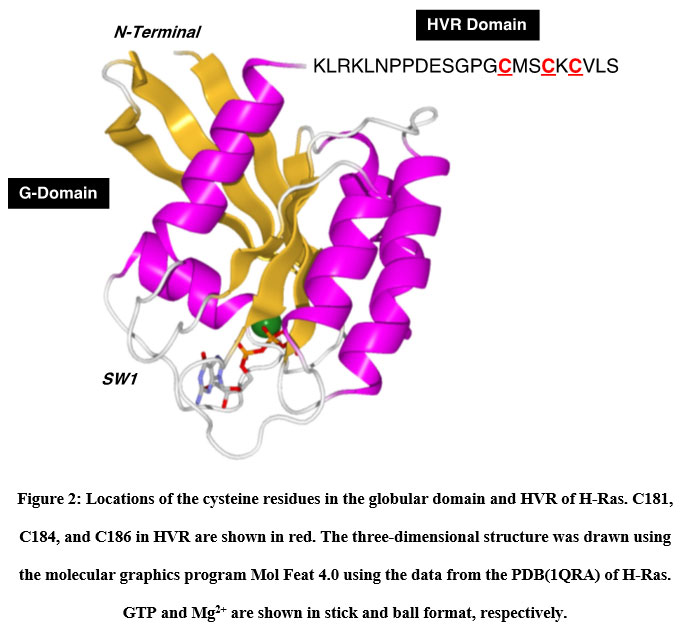 |
Figure 2: Locations of the cysteine residues in the globular domain and HVR of H-Ras. |
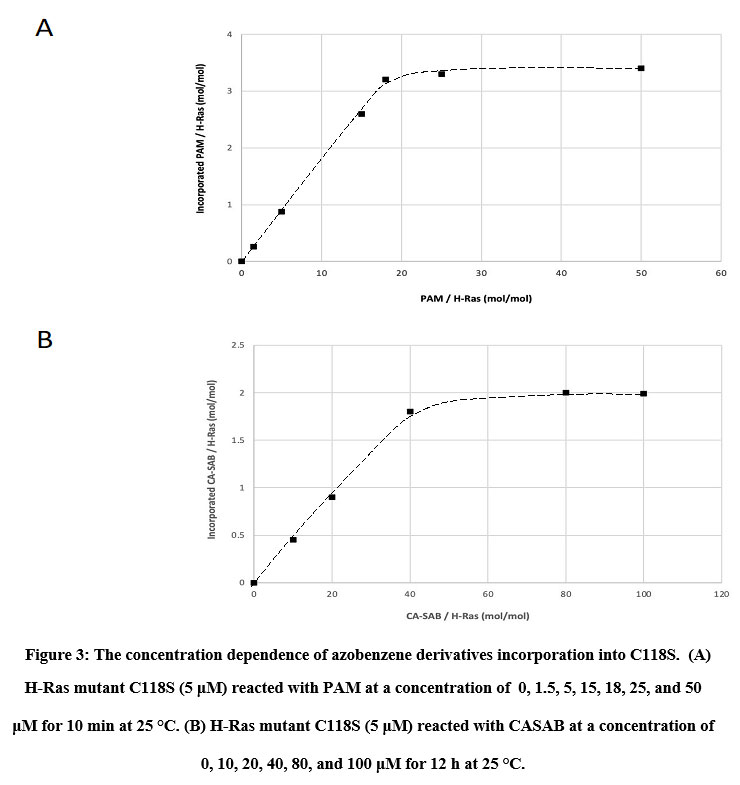 |
Figure 3: The concentration dependence of azobenzene derivatives incorporation into C118S. |
Photoisomerization of azobenzene derivatives incorporated into the HVR of H-Ras
The isomerization states, cis or trans, of azobenzene and its derivatives can be monitored using UV/visible light absorption spectroscopy 14. The absorption spectra of CASAB and PAM (data not shown) were almost identical and exhibited significant spectral changes upon UV and visible light irradiation, reflecting typical azobenzene cis and trans isomerization (Figure 4A, B). Free CASAB-and CASAB-modified H-Ras showed almost similar spectral changes to free PAM and PAM-modified H-Ras (data not shown), respectively, as shown in Figure 5. The results suggested that the azobenzene derivatives incorporated into H-Ras exhibited cis–trans isomerization.
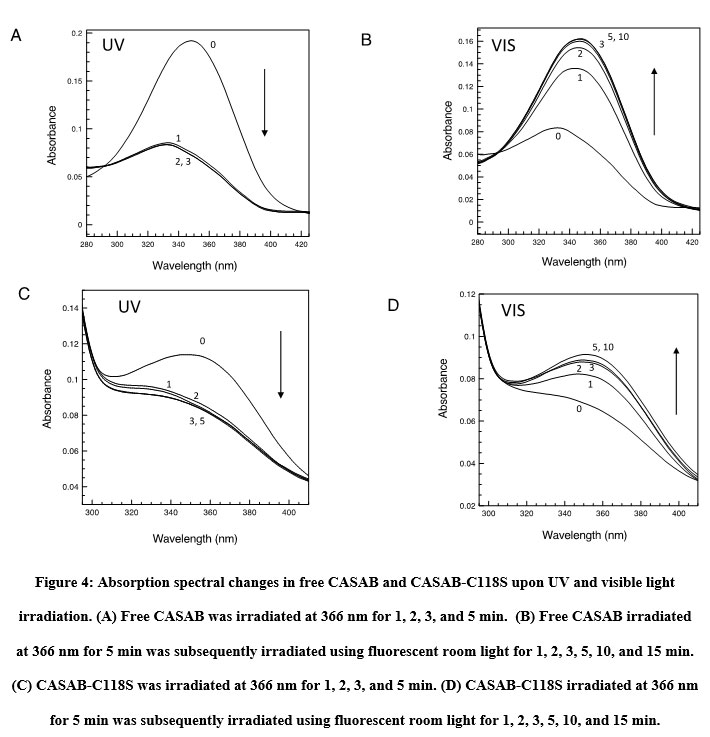 |
Figure 4: Absorption spectral changes in free CASAB and CASAB-C118S upon UV and visible light irradiation. |
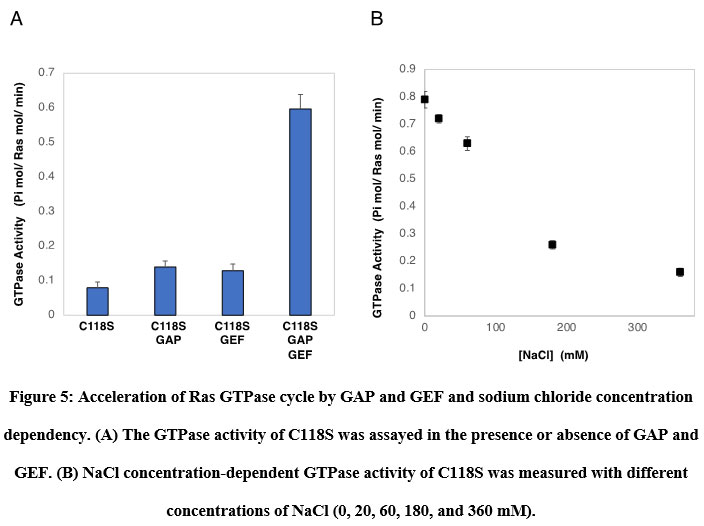 |
Figure 5: Acceleration of Ras GTPase cycle by GAP and GEF and sodium chloride concentration dependency. |
Ras GTPase cycle accelerated by GAP and GEF
Prior to examining the photoregulation of Ras GTPase by modification with photochromic compounds, we established a convenient Ras GTPase assay. It is known that intrinsic Ras GTPase activity switches to cellular signal transduction extremely slowly. However, it is not easy to show GTP hydrolysis in the catalytic site and monitor the exchange of GDP to GTP because of the requirements of highly sensitive detection methods using radioisotopes. We focused on the acceleration of the GTPase cycle by various factors. Ras continuously cycles GTPases in the presence of both active regulatory factors. As expected, in the presence of GAP and GEF, the GTPase of C118S was accelerated significantly by approximately 7.5 times, as shown in Figure 6A. Wild-type H-Ras showed almost identical GTPase activity (data not shown). GTPase activity was strongly dependent on the concentration of NaCl in the assay buffer (Figure 6B). The GTPase cycle accelerated by GAP and GEF produced sufficient Pi from GTP hydrolysis to be quantified using general Pi-detecting reagents.
Photocontrol of Ras GTPase by the isomerization of azobenzene derivatives incorporated into the HVR
The effect of isomerization of azobenzene derivatives bound to the HVR of Ras on the GTPase activity in the presence of GAP and GEF was examined. The GTPase activity of PAM- C118S was changed by UV and visible light irradiation (Figure 6A). The GTPase activities of trans-PAM-C118S and cis-PAM-C118S were 70% and 80%, respectively, compared with intact C118S. The differences in GTPase activity between the two isomerization states were not significant, but they showed reproducible photoreversible alteration upon three cycles of alternating UV and visible light irradiation. In contrast, CASAB-C118S changed its GTPase activity more significantly than PAM-C118S, accompanied by photoisomerization (Figure 6B). Trans-CASAB-C118S reduced GTPase activity to 62% of intact-C118S. Cis-CASAB-C118S exhibited almost the same GTPase activity (95%) as intact C118S. The GTPase activity of CASAB-C118S was reversibly controlled by UV and visible light twice with alternate irradiation (Figure 7).
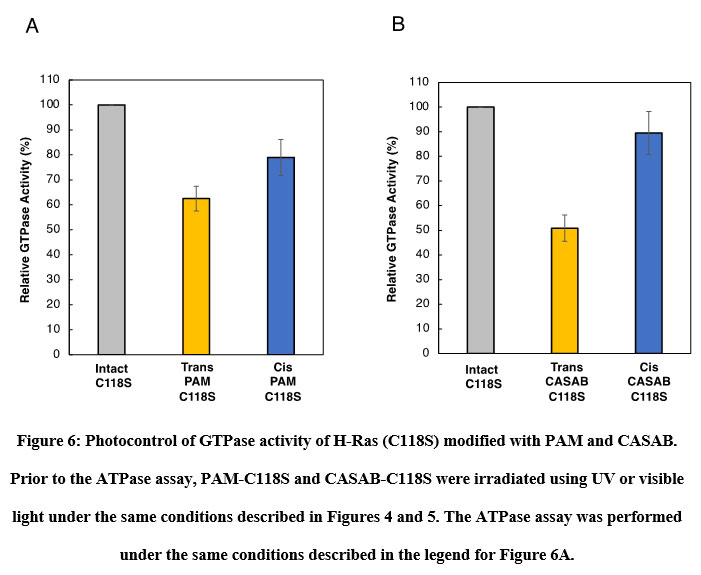 |
Figure 6: Photocontrol of GTPase activity of H-Ras (C118S) modified with PAM and CASAB. |
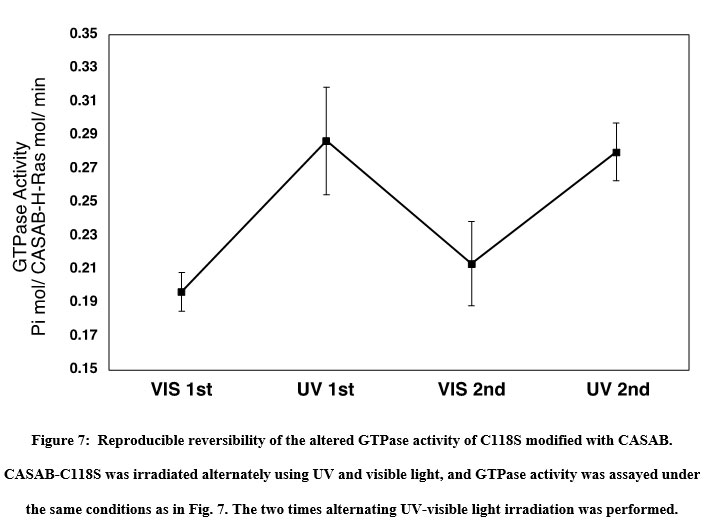 |
Figure 7: Reproducible reversibility of the altered GTPase activity of C118S modified with CASAB. |
Formation of H-Ras multimer by modification with azobenzene derivatives and its photocontrol
Formation of the Ras multimer induced by modification of HVR with PAM and CASAB was also examined using SEC-HPLC according to previously reported methods 11. As shown in Figure 8, the intact unmodified H-Ras mutant C118S eluted at approximately 7 min on the SEC-HPLC mentioned in the Materials and Methods section. PAM C118C showed an elution profile reflecting the formation of the Ras multimer, as we have previously observed 11. The peak at 5 min was consistent with the elution of the Ras multimer modified with caged compounds. Moreover, the cis isomer of PAM-C118S showed a slightly lower peak than the trans isomer (Figure 8). These results suggest that the cis and trans isomerization of PAM bound to the HVR lipidation sites changes the monomer-multimer equilibrium photo reversibly. In contrast, small broad peaks were observed at 5 min, while the cis and trans forms of CASAB-modified H-Ras exhibited almost no multimer formation (data not shown). This reflects the conformational differences in the HVR in the globular domain between the two isomerization states.
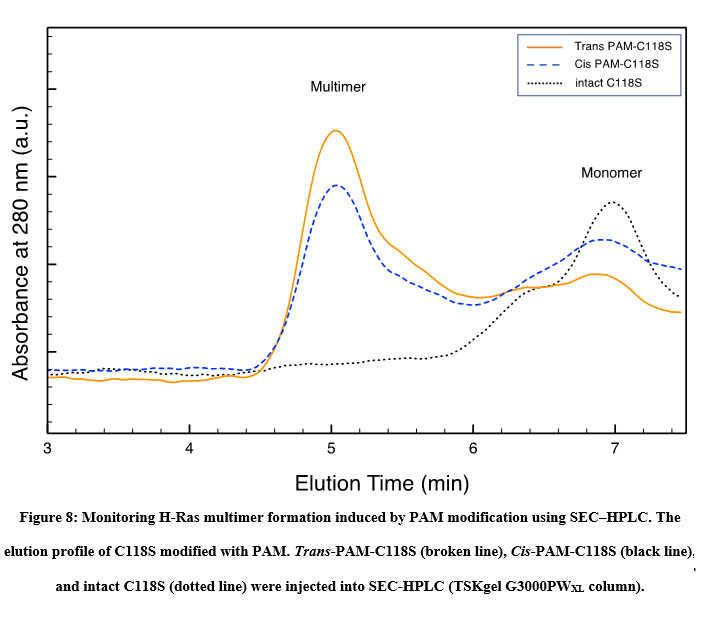 |
Figure 8: Monitoring H-Ras multimer formation induced by PAM modification using SEC–HPLC. |
Discussion
The aim of this study was to control the small GTPase Ras, which promotes cellular signal transduction using artificial external stimulus-responsive molecules. Previous crystallographic studies have suggested that the GTPase core part of small GTPases comprising Switch I, Switch II, and P-loop is strikingly similar to the ATP-driven motor proteins kinesin and myosin 15. Furthermore, structural analysis of the ATPase cycle 16-19 and the studies of point mutated motor proteins 20, 21 demonstrated a possible mechanical energy transduction system, suggesting that the ATP chemical energy is transmitted to physical-mechanical movement mechanically as a camshaft motion through rigid domain steric interactions. Therefore, nucleotide-driven functional proteins can be considered bio nanomachines. Furthermore, it is strongly expected that introducing photochromic molecules as artificial regulatory nanodevices into mechanically important sites enables the control of nanomachines as previous studies have suggested 22, 23. However, the direct incorporation of photo chromism into the catalytic region did not achieve highly efficient reversible control due to the direct interaction with the catalytic regions. Previously, Iwata et al. demonstrated that incorporating thiol-reactive caged compounds into HVR, one of the G protein regulatory domains, which regulates the physiological roles inducing multimer formation 8. However, the caged compounds exhibited irreversible photo regulation. Hence, we hypothesized that the incorporation of photochromic molecules into the HVR enables efficient control of the Ras function. In this study, we incorporated photochromic azobenzene derivatives into the HVR domain. HVR is the C-terminal region of the G protein consisting of three cysteine residues and a unique tail-shaped structure protruding from the catalytic globular domain as lipidation sites, leading to the formation of dimers or larger multimers on the plasma membrane, which is essential for signal transduction. Therefore, incorporating photochromic molecules into HVR as a photo switch is expected to exhibit highly efficient photo reversible regulation of Ras function related to physiological roles. In the present study, we utilized thiol-reactive azobenzene as a photo switching molecular device because of its drastic changes in molecular size and polarity by cis-trans isomerization. Therefore, it is expected that PAM and CASAB act as reversible photo switching nanodevices for Ras function. PAM, composed of a basic azobenzene moiety and a thiol-reactive maleimide group, is relatively hydrophobic. However, trans to cis isomerization drastically increases polarity. Therefore, the two isomerization states of PAM are expected to exert different influences on the HVR, which may act as a photo switching molecular device. As expected, PAM exerted photo regulation of H-Ras GTPase activity, as shown in Figure 7, and induced multimer formation, which may mimic physiological lipidation. Moreover, the cis isomer slightly decreased multimer formation (Figure 8). This may be due to the higher polarity of the cis isomer. Previously, we demonstrated that the formation of the Ras multimer induced by caged compounds strongly depends on ionic strength 11. Therefore, the driving force to form a multimer is considered to be electrostatic interaction, but not hydrophobic interaction with the modified HVR. Modifying HVR with hydrophobic aromatic compounds might induce conformational changes to open the electrostatic intermolecular interaction sites on the globular domain of H-Ras. The cis and trans isomers of PAM change the HVR conformation to induce slightly different states of the globular domain to interact intermolecularly, resulting in different efficient multimer formations. In contrast, H-Ras modified with CASAB did not form multimers (Figure not shown). CASAB contains a highly negatively charged sulfonate group, which is the complete opposite of hydrophobic lipidation. Therefore, modification with CASAB might not induce conformational changes to form multimers that mimic physiological clusters. However, although CASAB-H-Ras did not exhibit multimer formation, its GTPase activity was photo-controlled by cis and trans isomerization by UV and visible light irradiation more efficiently than PAM-H-Ras. As azobenzene drastically changes its structure by cis and trans isomerization 24, 25, the sulfonate moiety in CASAB changes its location on the HVR. Therefore, the trans isomer is thought to move the position of the sulfonate group to interfere with the GAP or GEF interaction with H-Ras, resulting in lower GTPase activity. The cis isomer exhibited almost normal GTPase activity.
Conclusion
We have successfully demonstrated that incorporating thiol-reactive photochromic azobenzene derivatives into the lipidation site in HVR, which is a physiologically functional site of H-Ras, enables the reversible control of GTPase activity and multimer formation by UV and visible light irradiation. The technique utilizing photochromic molecular devices may be applicable to other functional molecules, such as bio nanomachines. Further studies in this field are expected to contribute to the development of photo-regulated functional biomolecules with physiologically important roles.
Acknowledgment
We would like to thank Dr. Niitsu for helping with the FAB-MS analysis of the compounds used in this study.
Conflict of Interest
Authors declare no conflict of interests.
Funding Source
This work was supported by the JSPS KAKENHI Grant Grant-in-Aid for Scientific Research (C)15K07060.
References
- Fernandez-Medarde, A., & Santos, E. (2011). Ras in cancer and developmental diseases. Genes Cancer 2: 344–358.
CrossRef - Campbell, S. L., Khosravi-Far, R., Rossman, K. L., Clark, G. J., & Der, C. J. (1998). Increasing complexity of Ras signaling. Oncogene, 17(11), 1395-1413.
CrossRef - Marshall, C. B., Meiri, D., Smith, M. J., Mazhab-Jafari, M. T., Gasmi-Seabrook, G. M., Rottapel, R., … & Ikura, M. (2012). Probing the GTPase cycle with real-time NMR: GAP and GEF activities in cell extracts. Methods, 57(4), 473-485.
CrossRef - Kull, F. J., Vale, R. D., & Fletterick, R. J. (1998). The case for a common ancestor: kinesin and myosin motor proteins and G proteins. Journal of Muscle Research & Cell Motility, 19(8), 877-886.
CrossRef - Yamada, M. D., Nakajima, Y., Maeda, H., & Maruta, S. (2007). Photocontrol of kinesin ATPase activity using an azobenzene derivative. Journal of biochemistry, 142(6), 691-698.
CrossRef - Sadakane, K., Alrazi, I. M., & Maruta, S. (2018). Highly efficient photocontrol of mitotic kinesin Eg5 ATPase activity using a novel photochromic compound composed of two azobenzene derivatives. The Journal of Biochemistry, 164(4), 295-301.
CrossRef - Alrazi, I. M., Sadakane, K., & Maruta, S. (2021). Novel photochromic inhibitor for mitotic kinesin Eg5 which forms multiple isomerization states. The Journal of Biochemistry.
CrossRef - Iwata, S., & Maruta, S. (2015). Photocontrol of the GTPase activity of the small G protein K-Ras by using an azobenzene derivative. Biochemistry and biophysics reports, 4, 268-276.
CrossRef - Nussinov, R., Tsai, C. J., & Jang, H. (2019, February). Is Nanoclustering essential for all oncogenic KRas pathways? Can it explain why wild-type KRas can inhibit its oncogenic variant?. In Seminars in cancer biology(Vol. 54, pp. 114-120). Academic Press.
CrossRef - Rotblat, B., Belanis, L., Liang, H., Haklai, R., Elad-Zefadia, G., Hancock, J. F., … & Plowman, S. J. (2010). H-Ras nanocluster stability regulates the magnitude of MAPK signal output. PloS one, 5(8), e11991.
CrossRef - Nahar, R. (2021). Structural Analysis of Small G-Protein Ras Multimer Induced by Chemical Modification of HVR Domain with Caged Compound. Biophysical Journal, 120(3), 295a.
CrossRef - Ellis-Davies, G. C. (2020). Useful caged compounds for cell physiology. Accounts of Chemical Research, 53(8), 1593-1604.)
CrossRef - Shishido, H., Yamada, M. D., Kondo, K., & Maruta, S. (2009). Photocontrol of calmodulin interaction with target peptides using azobenzene derivative. Journal of biochemistry, 146(4), 581–590.
CrossRef - Rau, H. (2002). Photoisomerization of azobenzenes. Photoreactive Organic Thin Films, 3-47.
CrossRef - Kull, F. J., Vale, R. D., & Fletterick, R. J. (1998). The case for a common ancestor: kinesin and myosin motor proteins and G proteins. Journal of Muscle Research & Cell Motility, 19(8), 877-886.
CrossRef - Kull, F. J., Sablin, E. P., Lau, R., Fletterick, R. J., & Vale, R. D. (1996). Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature, 380(6574), 550-555.
CrossRef - Rayment, I., Rypniewski, W. R., Schmidt-Base, K., Smith, R., Tomchick, D. R., Benning, M. M., … & Holden, H. M. (1993). Three-dimensional structure of myosin subfragment-1: a molecular motor. Science, 261(5117), 50-58.
CrossRef - Fisher, A. J., Smith, C. A., Thoden, J., Smith, R., Sutoh, K., Holden, H. M., & Rayment, I. (1995). X-ray structures of the myosin motor domain of dictyostelium discoideum complexed with MgADP. cntdot. BeFx and MgADP. cntdot. AlF4. Biochemistry, 34(28), 8960-8972.
CrossRef - Dominguez, R., Freyzon, Y., Trybus, K. M., & Cohen, C. (1998). Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: Visualization of the pre–power stroke state. Cell, 94(5), 559-571.
CrossRef - Klumpp, L. M., Brendza, K. M., Gatial, J. E., Hoenger, A., Saxton, W. M., & Gilbert, S. P. (2004). Microtubule− kinesin interface mutants reveal a site critical for communication. Biochemistry, 43(10), 2792-2803.
CrossRef - Yun, M., Zhang, X., Park, C. G., Park, H. W., & Endow, S. A. (2001). A structural pathway for activation of the kinesin motor ATPase. The EMBO Journal, 20(11), 2611-2618.
CrossRef - Umeki, N., Yoshizawa, T., Sugimoto, Y., Mitsui, T., Wakabayashi, K., & Maruta, S. (2004). Incorporation of an azobenzene derivative into the energy transducing site of skeletal muscle myosin results in photo-induced conformational changes. Journal of biochemistry, 136(6), 839-846.
CrossRef - Ishikawa, K., Tamura, Y., & Maruta, S. (2014). Photocontrol of mitotic kinesin Eg5 facilitated by thiol-reactive photochromic molecules incorporated into the loop L5 functional loop. The Journal of Biochemistry, 155(3), 195-206.
CrossRef - Timpe, H. J. (1991). Photochromism–molecules and systems. Herausgeber: Dürr, H., Bouas‐Laurent, H. 1. Auflage, 1068 S. Amsterdam, Oxford, New York, Tokyo: Elsevier, 1990. Schriftenreihe: Studies in organic chemistry, 40. ISBN 0‐444‐87432‐1.
CrossRef - Beharry, A. A., & Woolley, G. A. (2011). Azobenzene photoswitches for biomolecules. Chemical Society Reviews, 40(8), 4422-4437.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.



