

How to Cite | Publication History | PlumX Article Matrix
Abdelmajeed M. Elshafei1 , Nuha A. Mahmoud1 , Yassir A. Almofti1,2*
, Nuha A. Mahmoud1 , Yassir A. Almofti1,2*
1Faculty of Medicine and Surgery, National University/Sudan
2Department of Molecular Biology and Bioinformatics, College of Veterinary Medicine, University of Bahri, Khartoum- Sudan.
Corresponding Author E-mail: yamofti99@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3032

ABSTRACT: Background: Human papillomavirus 16 (HPV16) is a small non-enveloped DNA virus is belonging to Papillomaviridae. It usually causes warts and about 60% of cancer diseases. HPV16 genome consists of double-stranded cDNA of six early and two late proteins. This study attempted to design safe and efficient multi epitopes vaccine from structural proteins (L1 and L2) by using various immunoinformatic databases. The results demonstrated that the predicted vaccine comprised of 408aa and validated in terms of antigenicity, allergenicity, toxicity and stability by putting all critical parameters into consideration. The physiochemical properties displayed isoelectric point (pl) of 10.37. The instability index (II) was 33.6 categorizing vaccine as stable. The aliphatic index was 63.24 and the GRAVY was −0.652 demonstrating the hydrophilicity of the vaccine. Vaccine structures were predicted, refined and validated. Stability of the vaccine was assessed through Ramachandan plot and further assessed by ProSA server. Vaccine solubility was higher than the solubility of E. coli proteins indicating that the vaccine was soluble. Disulfide engineering increased the vaccine stability by substituting the unstable residues with cysteine residues. Vaccine-TLR4 receptor docking resulted in attractive binding energy of –1274.1 kcal/mol and –1450.4kcal/mol for chain A and chain B of the receptor respectively. Reverse transcription of the vaccine protein into a DNA sequence was performed and cloned into a pET30a (+) vector to confirm the clonability of the sequence during microbial expression. Taken together, the vaccine potentially induced immune responses and thus was suitable as a vaccine to combat HPV16 disease. Nonetheless, the efficiency of vaccines must be approved by in vitro and in vivo immunological analysis.
KEYWORDS: Epitopes; Immunoinformatics; Human Papilloma Virus-16; L1; L2
Download this article as:| Copy the following to cite this article: Elshafei A. M, Mahmoud N. A, Almofti Y. A. Development of Multi-Epitopes Vaccine against Human Papilloma Virus16 Using the L1 and L2 Proteins as Immunogens. Biosci Biotech Res Asia 2022;19(3). |
| Copy the following to cite this URL: Elshafei A. M, Mahmoud N. A, Almofti Y. A. Development of Multi-Epitopes Vaccine against Human Papilloma Virus16 Using the L1 and L2 Proteins as Immunogens. Biosci Biotech Res Asia 2022;19(3). Available from: https://bit.ly/3DSdgkG |
Introduction
The Human papillomavirus (HPVs) is a viral infection that belongs to the Papillomaviridae family consists of small, non-envelope DNA viruses commonly cause skin or warts. The genome consists of double-stranded cDNA that decodes for an early six proteins (E1, E2, E4-7) and two late proteins (L1 and L2) 1. Virus replication and translation are primarily dependent on the E1 and E2 proteins [1]. The expression of E4 and E6 is also regulated by E2. E5 is essential for viral assembly and growth. Major (L1) and minor capsid (L2) proteins are structural proteins [1]. HPV is composed of quite 100 varieties that mainly infect the cervix and the oropharynx. According to their oncogenic potential, the types of HPV are often categorized as high-risk or low-risk 2. The HPV16 virus is regarded as having the greatest potential for causing cancer [2]. Among all human cancers, 15% are caused by viral infections. Approximately 600,000 cases of cervix cancer, oropharyngeal, anal, vulvovaginal and penile cancer, as well as recurrent papillomatosis of the lungs have been associated with HPV 3. HPV infects both the cutaneous and mucosal squamous epithelium exclusively through intraepithelial transmission. Among the HPVs that cause cancer, HPV type 16 and type 18 are the most common 4. Among a minimum of 13 genotypes of high-risk HPVs, HPV16 is highly frequent in cervical cancer, demonstrating 60% of the cases 5. HPV16 also frequent in other oropharyngeal and anogenital cancers 6. The oropharynx squamous cell carcinomas (OPSCC) of the oropharynx, for example, are more common than lymphomas, sarcomas and cancers of the minor salivary glands 6. In the past, tobacco smoking and alcohol abuse were primary risk factors for OPSCC. Nevertheless, this has declined in recent years, reflecting lower tobacco consumption rates in the United States 7. Over the same time, oropharyngeal cancers due to infection with high-risk HPV (HPV16) have been increasingly recognized. HPV-related OPSCCs have increased by 225% in the past three decades while tobacco-related OPSCCs have declined 8. HPV16 plays an important biological and clinical role in HPV-associated malignancies 8.
HPV types can be distinguished by quite a few differences within the sequence of the L1 capsid gene compared to other types 9. Variability exists among each type of HPV, with differences of 1-10% and 0.5-1% in full sequence categorized as variant lineage and sub-lineage, respectively. Therefore, HPV16 comprises four lineages and a minimum of nine sublineages 10. In addition, each lineage/sublineage has an unusually high number of single nucleotide polymorphisms.
Recently, the preventive HPV vaccination based on L1, has a compelling safety profile as well as clinical effectiveness against the HPV genotypes from which it was deprived 11. These continued efforts were undertaken in the field of L1-based vaccinations to improve their efficacy by broadening the scope of protection and lowering the cost of these vaccines for greater access and effective prevention of HPV infections 11. Cervarix vaccine has already been approved by some countries and is under review by the FDA 12. Cervarix uses L1 as an immunogenic factor, which contains HPV types 16 and 18, the two primary serotypes linked to cervical cancer. As a result, the vaccination has been designed to target two major cancer-causing strains of HPV (16 and 18), which can be over 70% of all cervical malignancies 12. However it has been developed to protect only virus infection from HPV types 16 and 18 (restricted protections). This vaccine is in the clinical trial stage (phase III) test. The clinical study of cervarix in healthy volunteers of various ages revealed increased antibody levels in the pre-teen/adolescent group compared to women 15–25 years old 13. Although the Cervarix duration protection is not yet known, newly available data showed that Cervarix is particularly effective against HPV16/18 for up to 5.5 years and avoids the majority of CIN2+ lesions 14. In addition, the vaccination demonstrated ongoing cross-protection against HPV45 and HPV31 incident infections 14.
This study aimed to design multi epitopes vaccine for HPV16 by using immunoinformatic databases from L1 and L2 structural proteins to act as safe and efficient vaccine without future complication.
Material and methods
Viral proteins retrieval and sequence alignment
The whole proteome of HPV16 was available in the National Center For Biotechnology Information (NCBI) at (https://www.ncbi.nlm.nih.gov/protein). The virus provided eight proteins. Table (1) provided the names, accession numbers, amino acids lengths and functions of each protein.
Table 1: HPV16 entire proteins assembly with accession number, length, and antigencity.
|
Viral protein |
Accession no |
Function |
Length |
aVaxijen antigenicity |
|
L1 |
NP_041332.2 |
Major capsid protein |
505aa |
0.5150 ( antigenic ) |
|
L2 |
NP_041331.2 |
Minor capsid protein |
473aa |
0.6457 (antigenic) |
|
E1 |
NP_041327.2 |
Viral protein replication |
649aa |
0.4427 (antigenic) |
|
E2 |
NP_041328.1 |
Viral protein replication: repression of E6/E7 gene |
365aa |
0.4279 (antigenic) |
|
E4 |
Yp_009268708.1 |
Assembly & release of viral particle |
92aa |
0.4369 (antigenic) |
|
E5 |
NP_041330.2 |
Interaction with epidermal growth factor (EGF) |
83aa |
0.3507 ( non-antigenic ) |
|
E6 |
NP_041325.1 |
Destruction of p53 tumer suppression protein |
158aa |
0.6921 (antigenic) |
|
E7 |
NP_041326.1 |
Inactivation of pRb tumers suppress protein |
98aa |
0.5765 (antigenic) |
athe cut off value for the Vaxijen antigenicity was the default value (0.4)
Physical and chemical features, transmembrane topology and the antigenicity of the viral proteins
The Expasy Prot Param server at (http://web.expasy.org/protparam/) was exploited to compute various physiochemical features for multiple protein sequences. The server was used to compute physiochemical features for each protein sequence of HPV16 such as the molecular weight, amino acid composition, instability index and gravy values etc.). The viral proteins were further examined for transmembrane topology and antigenicity using TMHMM server at (http://www.cbs.dtu.dk/services/TMHMM/) and Vaxijen v2.0 server at (www.ddg-pharmfac.net/vaxijen/), respectively. A set of proteins had been chosen for further analysis based on their physicochemical features, transmembrane topologies and antigenicity. Accordingly, as shown in Table (1): the late structural proteins (L1 and L2) demonstrated good physiochemical features and antigenicity. These two structural proteins were elected for the prediction of epitopes to act as vaccine candidates.
Strains retrieval of L1 and L2 and epitopes conservancy
A total of 165 and 12 strains sequences were retrieved from the NCBI for L1 and L2 proteins, respectively. These strains were aligned via multiple sequence alignment (MSA) based on the protocol of Clustal W, presented in the BioEdit software, version (7.0.9.0) 15. The conserved predicted epitopes were obtained from the MSA of the retrieved strains.
Prediction of the B-cells interacting Epitopes
Immune cells recognize the antigenic determinants as parts of the antigen that bind to B lymphocytes. They played a vital role in vaccine design 16. The ABCpred server (https://webs.iiitd.edu.in/raghava/abcpred/ABC_method.html) was exploited to predict B cell epitope with the threshold of 5.1 and epitopes length of 12mers from L1 and L2 protein. A multiple sequence alignment of protein strains, as well as predictions of B cells epitope conservation, was performed with Bioedit software 15.
Interaction of Major Histocompatibility Complex-1 (MHCI) Epitopes with cytotoxic T lymphocytes
This prediction was assessed via Immune Epitopes Data Base (IEDB) MHC-I tools at (http://tools.iedb.org/mhci/). There were multiple steps involved in the MHC-I epitopes interactions with cytotoxic T lymphocytes. For instance, the artificial neural network 4.0 (ANN 4.0) method was used as a prediction method, the length of epitopes was set to nine amino acids and the percentile rank of ≤ 1 was used for the allele’s epitopes interaction 17,18.
Interactions of Major Histocompatibility Complex-II (MHCII) Epitopes with Helper T lymphocytes
This prediction was assessed via IEDB MHC-II prediction tool at (http://tools.iedb.org/mhcii/result/). Peptides were analyzed for their ability to interact with helper T lymphocytes using the human HLA-DR, HLA-DP, and HLA-DQ reference allele sets. Due to the fact that the MHC-II groove can bind to various lengths, the neural network align 2.3 (NN-align 2.3) method was employed to determine both the binding affinity of the peptides and the core epitopes to MHC-II. The peptide length was adjusted to18 amino acids. The percentile rank of ≤10 was used for the allele’s epitopes interaction 19.
Epitopes antigenicity, allergenicity, and toxicity prediction
Several prediction tools were used to analyze whether the predicted epitopes were antigenic, allergenic, and / or toxic. Antigenicity of predicted epitopes was determined using Vaxijen v2.0 server at (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). The default threshold of Vaxijen server was used (0.4). AllerTOP server was used to examine allergenicity 20 and ToxinPred server was used to determine the toxicity of epitopes 21.
Population coverage analysis
The interacting epitopes from MHC-I and MHC-II molecules were examined for population coverage against the whole world after they were proved to be antigenic, nonallergic, and non-toxic. The analysis was performed by using the population coverage tool from IEDB (http://tools.iedb.org/ tools/population/iedb_input).
Vaccine Construction
The vaccine construct was created from the epitopes elected as B-cell epitopes as well as epitopes that had a high level of allelic interaction against T lymphocytes. GPGPG sequence was exploited to fuse B cells and cytotoxic T cells epitopes. The KK linker was used to bind T-helper cell epitopes. Human β-defensin 3 (UniProt entry Q5U7J2) was added to the vaccine sequence at the amino terminal as an adjuvant to improve vaccine immunogenicity 22. Moreover, β-defensin has been found to elicit immunogenic responses similar to those in the innate immune system 22. The adjuvant was separated from the vaccine sequences by EAAAK linker. Also, linkers have been found to augment protein stability by separating its functional domains 23,24.
The Physiochemical properties of the designed vaccine
The Protparam analysis tool (https://web.expasy.org/protparam/) was used to analyze the physical and chemical attributes of the designed vaccine. Among the parameters computed were the amino acid composition, molecular weight, instability index, theoretical isoelectric point (pI) and the Grand average of hydropathicity index (GRAVY).
Homology assessment of the vaccine to human proteome
The homology assessment of the vaccine protein to human whole proteins was applied via NCBI BLASTp 25, 26. Homology analysis aimed to prevent autoimmunity caused by the similarity between human proteins and vaccines. Protein BLASTp search was constricted to records of Homo sapiens taxid No: 9606. Sequence homology must fall below 40% in homology to the human proteome 27.
Prediction of the vaccine secondary structure
SOPMA server at (http://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?p=NPSA/npsa_sopma.html) was used to predict the vaccine secondary structure 28.
Prediction, refinement, and validation of vaccine tertiary structures
Raptor X server was used to predict vaccine 3D structure after submission of the vaccine primary sequence to the server 29. The server donates the correct prediction of the protein function and structure 29. The PDB file provided by raptor X server was submitted to GalaxyWEB server for refinement 30. Protein refinement was used to improve the physical quality of the vaccine structure. Ramachandran plot embedded at Saves v6.0 server at (https://saves.mbi.ucla.edu/) was carried out to assess the stability of the refined protein structure. Additionally, the refined PDB file was examined by ProSA server and the result was provided as Z score for possible errors in the structure 31.
Determination of solubility and stability properties of the vaccine
Vaccine solubility was analyzed by Protein-sol server 32. This web service offers mathematical and predictive methods for determining the solubility of proteins 32. The scaled query solubility value of the vaccine (QuerySol) was analyzed versus the E. coli experimental population datasets (PopAvrSol of 0.45). Thus solubility scores larger than 0.45 indicated that the protein is more soluble than the average E. coli solubility 33,34. Moreover, the disulfide bonding provided the vaccine structure a stronger geometric conformation and made it more stable. The tool used for disulfide engineering in vaccine construct was the Disulfide by Design 2.0 (DbD2) software 35. To predict disulfide bonds in a protein structure model, all residue pairs must be evaluated for distance and geometries that enable disulfide to occur, assuming that cysteine substitutes these residues 35.
Molecular docking using TLR4 receptor for constructed vaccine
Several biological processes depend on protein–protein and protein–DNA/RNA interactions could be used for molecular complex docking. Cluspro server at (https://cluspro.bu.edu/login.php?redir=/queue.php) uses protein-protein docking process based on three computational steps. Initially: the server performs rigid body docking by scanning billions of conformations. Secondly: RMSD-based clustering of 1000 lowest energy structures to find the largest cluster representing the most probable model of the complex. Thirdly: elimination of energy used in space collisions minimizing the docking of the ligand (vaccine) to receptor 36. The protein data bank (PDB) file number (4G8A) of the toll like receptor 4 (TLR4) was used as the receptor of the docking process with the vaccine construct PDB file as a ligand. The docking interaction was visualized by pymol server at (www.pymol.org).
In silico cloning
This was performed to assess the translational expression of the vaccine in E. coli strain K12 after reverse transcription of the vaccine protein into a DNA sequence using the Java Codon adaptation server (JCAT) at (http://www.prodoric.de/JCat). The Rho-independent termination of transcription, the prokaryotic ribosome binding site and the restriction enzyme cleavage site of the server were avoided 37. In JCAT server, firstly the codon adaptation index (CAI) in the server ranging from 1 to 0.8 is considered better with favorable GC content range from (30-70%) [38]. Secondly, the DNA sequence obtained via the server (JCat) was supported by linking of restriction enzyme cleavage sites of BamHI and Xho1at the ends of the DNA sequence. The DNA sequence was then inserted into pET30a (+) vector using SnapGene software between BamHI and Xho1 restriction enzymes 37-39.
Results
Sequences alignment
ClustalW was used to align all the obtained strains using the Bioedit software. Figure (1) demonstrated the alignment of L1 and L2 strain sequences. Each variant was checked for epitopes conservancy. Mutated epitopes were considered non-conserved epitopes and were excluded while non-mutated epitopes were considered conserved epitopes and were used for further investigation.
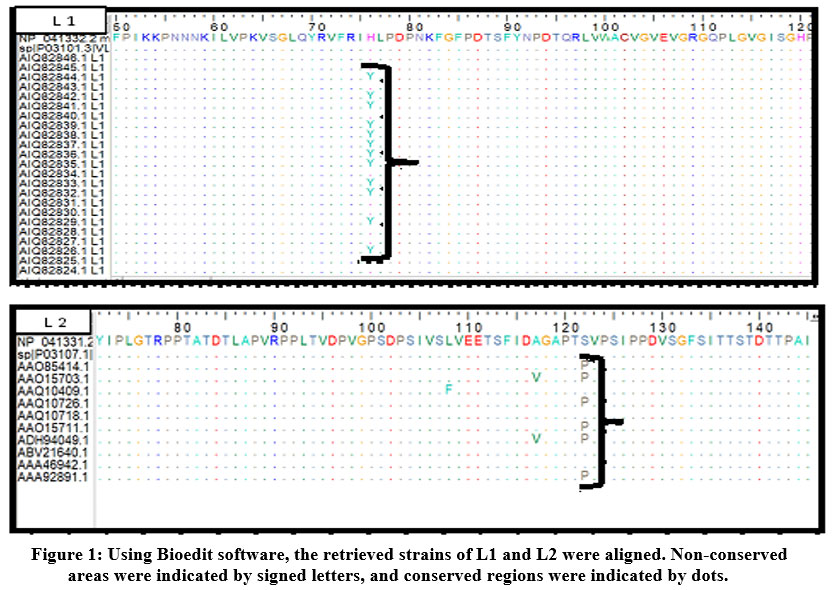 |
Figure 1: Using Bioedit software, the retrieved strains of L1 and L2 were aligned. Non-conserved areas were indicated by signed letters, and conserved regions were indicated by dots. |
B-cell epitopes prediction
The sequences of L1 and L2 proteins were submitted to the ABCpred server. B cell epitopes were graded and predicted based on their scores by a trained recurrent neural network. The greater the score, the higher the probability of B cell epitopes. All the results of B cells peptides were above the 0.51 threshold value. Then the predicted B cell epitopes were examined for their conservancy with the BioEdit tool, and also tested for antigenicity, allergenicity, and toxicity. The antigenic, non-allergic and non-toxic B cells epitopes are shown in table (2) and further selected to assemble the multi epitopes vaccine.
Table 2: B cell selected epitopes as vaccine candidates from L1 and L2 proteins.
|
Protein |
Epitopes |
score |
Start |
End |
Vaxijen antigenicity |
Allergenicity |
Toxicity |
|
L 1 |
VGRGQPLGVGIS |
0.78 |
107 |
118 |
1.8334 |
Non allergic |
Non-toxic |
|
|
VEVGRGQPLGVG |
0.63 |
105 |
116 |
1.8750 |
Non allergic |
Non-toxic |
|
|
ECISMDYKQTQL |
0.62 |
145 |
156 |
1.8412 |
Non allergic |
Non-toxic |
|
|
GRGQPLGVGISG |
0.6 |
108 |
119 |
1.4764 |
Non allergic |
Non-toxic |
|
|
ACVGVEVGRGQP |
0.59 |
101 |
112 |
1.5264 |
Non allergic |
Non-toxic |
|
L 2 |
GFSITTSTDTTP |
0.78 |
132 |
143 |
0.9047 |
Non allergic |
Non- toxic |
|
|
TGGRTGYIPLGT |
0.75 |
66 |
77 |
1.4318 |
Non allergic |
Non- toxic |
|
|
GLYSRTTQQVKV |
0.73 |
226 |
237 |
0.9552 |
Non allergic |
Non- toxic |
|
|
RPPLTVDPVGPS |
0.71 |
90 |
101 |
0.5871 |
Non allergic |
Non- toxic |
|
|
VALHRPALTSRR |
0.71 |
287 |
298 |
0.7737 |
Non allergic |
Non- toxic |
The threshold for the Vaxijen antigenicity was 0.4
CTL epitopes prediction
Multiple epitopes were predicted to interact against cytotoxic T cells from L1 and L2 proteins using IEDB MHC-I binding prediction tools. Five epitopes from L1 protein and four epitopes from L2 proteins were shown to be conserved epitopes with antigenicity, non-allergenicity and non-toxicity. Moreover, these epitopes provided high allelic interactions with MHC-I alleles and high population coverage scores. Based on these criteria they were elected as vaccine candidates and are shown in table (3).
Prediction of T helper- lymphocyte epitopes
The IEDB MHC class II binding prediction tool was used to analyze L1 and L2 proteins to provide T helper cell epitopes interacting with MHC class II alleles (HLA-DQ, HLA-DR and HLA-DP). A number of conserved, antigenic, nonallergenic and nontoxic epitopes were predicted. Based on the allelic interaction and high population coverage four epitopes from each protein were selected (table 4).
Table 3: Cytotoxic T cells selected epitopes as vaccine candidates from L1 and L2 proteins with their population coverage scores.
|
Protein |
Epitopes |
Start |
End |
Vaxijen antigenicity |
Population coverage |
|
L1 |
TTSSTSTTA |
490 |
498 |
0.7108 |
20.09% |
|
|
VEVGRGQPL |
105 |
113 |
1.2418 |
27.36% |
|
|
WEVNLKEKF |
447 |
455 |
1.4265 |
20.05% |
|
|
YDLQFIFQL |
370 |
378 |
1.6395 |
27.36% |
|
|
YPDYIKMVS |
231 |
239 |
0.4994 |
55.20% |
|
L2 |
EIPMDTFIV |
195 |
203 |
0.8318 |
75.86% |
|
|
KRRKRLPYF |
457 |
465 |
1.7971 |
67.59% |
|
|
MLRKRRKRL |
454 |
462 |
0.7552 |
90.96% |
|
|
YLHPSYYML |
447 |
455 |
0.9664 |
98.80% |
The vaxijen antigenicity threshold was the default threshold of the server (0.4). All the predicted epitopes were shown to be nonallergen and nontoxin in allerTOP and toxinpred servers
Table 4: Helper T cells selected epitopes as vaccine candidates from L1 and L2 proteins with their population coverage scores.
|
Protein |
Core peptide |
Peptide |
Start |
End |
Vaxijen antigenicity |
population coverage |
|
L 1 |
ATPTTSSTS |
EDPLKKYTFWEVNLKEKF |
438 |
455 |
0.6432 |
76.04% |
|
|
ATVYLPPVP |
KEDPLKKYTFWEVNLKEK |
437 |
454 |
0.4953 |
81.18% |
|
|
FWEVNLKEK |
KKYTFWEVNLKEKFSADL |
442 |
459 |
1.7566 |
67.14% |
|
|
GICWGNQLF |
LEDTYRFVTSQAIACQKH |
414 |
431 |
0.9516 |
63.39% |
|
L 2 |
FFGGLGIGT |
TSFIDAGAPTSVPSIPPD |
112 |
129 |
1.0853 |
70.96% |
|
|
ILQYGSMGV |
FIVSTNPNTVTSSTPIPG |
201 |
218 |
1.2878 |
60.05% |
|
|
LHPSYYMLR |
PDFLDIVALHRPALTSRR |
281 |
298 |
0.9761 |
90.39% |
|
|
YLHPSYYML |
DFYLHPSYYMLRKRRKRL |
445 |
462 |
0.9664 |
79.19% |
The vaxijen antigenicity threshold was the default threshold of the server (0.4). All the predicted epitopes were shown to be nonallergen and nontoxin in allerTOP and toxinpred servers
Physiochemical properties of the vaccine
The proposed vaccine was a combination of nine cytotoxic T cell epitopes, ten B-cell epitopes, and eight helper T cell epitopes (figure 2). The adjuvants and linkers were added to improve the vaccine immunogenicity. The vaccine was antigenic in Vaxijen server (0.8534) and nonallergen. The molecular weight determined by (Protparam server) was 44.4 kilo Dalton and the pI was 10.37 indicating the vaccine was alkaline. Negatively and positively charged residues were 18 and 78, respectively. Instability index (II) score was 33.6 categorizing the vaccine as stable. Aliphatic index was 63.24 and the GRAVY was −0.651 showing the hydrophilicity of the vaccine.
 |
Figure 2: The residues of the multi-epitope vaccine. Helper T and B cells predicted epitopes were linked via the KK linkers, whereas cytotoxic T epitopes were linked via the GPGPG linkers. |
Homology assessment by BLASTp
BLASTp homology assessment was performed to assess whether the vaccine would be implicated in autoimmune diseases to the host’s health. It was determined that only 11% of the vaccine proteome sequences were homologous to human whole proteins indicating that the vaccine does not compromise the host’s health.
Secondary and tertiary structure of vaccine
The vaccine predicted secondary structure was provided in figure (3). The vaccine showed 8.33%, 9.8% and 28.6% as α-helix, β-turn and extended strands, respectively. The random coiled residues scored 53.19%. The 3D structure of the vaccine was predicted by Raptor X server, refined with the Galaxy refiner server (figure 4a, 4b). The Ramachandran plot was used to evaluate the refined 3D structure. The plot showed 83.7% of the residues in the favoured region, 14.6% of the residues in the allowed region and only 1% of the residues in the outlier region (figure 4c). Moreover, the good quality of the model was proved by the Z-score of – 4.46 in the ProSA server (figure 4d).
 |
Figure 3: The 2D structure prediction of the vaccine. Alpha helices, extended strands and beta turns were shown in blue, red and green colors, respectively. |
 |
Figure 4: (a) The 3D model of the vaccine (b) the refined 3D model. (c) The validated or evaluated model by Ramachandran plot. (d) Z-score of −4.46 (ProSA-server). |
Solubility and stability of the vaccine
The solubility score of the vaccine was 0.636 contrasted to 0.45 of the population average solubility of E. coli (figure 5). This result indicated the vaccine was soluble. For stability, from 30 pair residue with disulfide bonds, only five pairs of residues could be replaced with cysteine. Those pairs were (34ser -39lys), (112met-115lys), (187asp-190gly), (204ser-207lys) and (352glu-377gly) (figure 6).
 |
Figure 5: The vaccine solubility score was (0.636) greater than that of the population average solubility of E. coli (0.45). |
 |
Figure 6: vaccine stability in the original form (left). Five golden sticky forms (disulfide bond regions) indicated by red arrows in the mutant form (right fig). |
Molecular docking
Molecular protein-protein docking was performed with CLUSPRO docking server to find the binding score of vaccine against toll like receptor 4 (TLR4) chains. The designed vaccine showed favourable interaction with TLR4 shown in figure (7). Docking of the TLR4 with vaccine showed efficient binding energy of –1274.1 kcal/mol and – 1450.4 kcal/mol for chains A and B, respectively.
 |
Figure 7: Docking of the vaccine against TLR4 chains represented by green and brown colors respectively. |
Codon adapting and in silico cloning
The vaccine sequence was reverse transcriped into DNA sequence. CAI Value was 0.95, indicating the high abundant codons proportion. Vaccine DNA sequence demonstrated favourable GC content (69. 4 %). Figure (8) showed cloning of the DNA sequence into pET30a (+) vector between BamHI and Xho1 restriction enzymes cutting sites.
 |
Figure 8: cloning of the vaccine (red colour) into the pET30a (+) vector (black colour). The DNA sequence was successfully cloned between the BamH1 and Xho1 of the vector. |
Discussion
Bioinformatics tools have led to significant time and resource savings in vaccine research in recent years. These tools aid in the development of a multi-epitopes vaccine by identifying antigenic domains. The genomic and proteomic information about different viral pathogens has been enhanced through the advancement of sequence-based technologies 40. In this study, the bioinformatics tools were used to help in designing peptide vaccines using neutralizing epitopes based on various bioinformatics tools. An in silico epitope-based vaccine was implemented for many viral diseases, for instance, human immunodeficiency virus (HIV) 41,42, coronavirus 43 dengue virus 44 and viral encephalitis of Saint Louis 45.
Concerning the HPVs, the multi-epitopes DNA vaccine designed by Gupta and his colleagues used consensus epitopes sequences present in L2 protein of HPVs [46]. In addition, bioinformatics tools were used to enhance the immunogenicity of DNA vaccines by engineering CpG motifs in HPV genome 46. Hasseini and colleagues identify peptide vaccine that would protect against HPV type 11, 16, 18, 31, and 45. They performed an in silico examination of L1 and L2 protein to these types 47. For identification of T-cell CD8+ epitopes, Singh (2016) tested E1, E2, E6 and E7 high-risk proteins of HPV types for vaccination. The analysis protected high-risk HPV types and 14 epitopes were recommended from the HPV proteome 48. For identification of antigenic epitopes to induce the immune system against HPV16, 18, 31, and 45, Panahi and his co-workers used a two-step model including a sequence-based approach and molecular docking 49. Compared to these mentioned studies, the viral whole proteins of HPV16 were obtained from the NCBI database in this study. The antigenicity of each protein was assessed via Vaxijen server particularly for the structural proteins (L1 and L2).
Vaccine design is a complex matter with various factors taken into account; the most essential one is the safety and effectiveness of the vaccine 50. The allergenicity and toxicity of elected epitopes were considered to ensure the safety of the proposed vaccine 51, 52. The immunogenicity of antigen (vaccine), amino acids solvent accessibility, B cells recognition and binding to MHC molecules were also considered to ensure the effectiveness of the predicted epitopes 53,54. Thus the major and minor capsid proteins of HPV16 were subjected to ABCpred server to obtain B cells epitopes. Epitopes with 100% conservancy underwent further analysis to be antigenic, non-allergenic and non-toxic. For B cells, ten epitopes were shown to be antigenic, non-allergic, and nontoxic and thus chosen to enter the vaccine structure. Also each of the reference sequences of the L1 & L2 proteins were examined using IEDB MHC-I and MHC-II prediction methods for T cell epitopes bound with MHC class I and class II alleles. The MHC-I and the MHC-II epitopes from both the L1 and L2 proteins demonstrated high binding score to MHC-I and MHC-II alleles, had favourable antigenicity score by Vaxijen server, and they were shown to be non-allergic and non-toxic, hence were selected to enter in the vaccine structure.
The assembled vaccine structure was made with the aid of suitable protein spacers or linkers. Linkers have displayed an increased significance in the assembly of stable, bioactive fusion proteins 55. If functional domains are directly fused without a linker, many adverse outcomes may occur, including protein misfolding 56, decreased rate of protein production 57, or diminished bioactivity 58. The linkers allowed the creation of a sequence with minimal junctional immunogenicity 16. In addition, the vaccine was enhanced by the addition of the adjuvant at the N-terminus of the construct. Linker EAAAK was used to control the distance and reduce interference between the adjuvant and the vaccine domains at a high level of expression 59,16. In this study, the β-defensin was exploited as an adjuvant for its relatively small size (45 amino acids) as well as its capability to perform as an immunomodulator and antimicrobial agent 60. Finally, a six histidine-tag sequence was linked at the carboxyl terminal of the vaccine. The small size of the His-tag would not alter the protein structure which is useful for downstream assays, purification and the ease defining of the protein function 61,62.
This chimeric construct was then assessed for the physical and chemical, immunological and structural features via different bioinformatics tools to endorse the validity and potency of the construct. By using the ProtParam server; the physicochemical properties classified the vaccine as a stable protein with potential hydrophilicity. To ascertain the antigenicity and allergenicity of this construct; it was assessed by VaxiJen and AllerTOP servers to show up non-allergenicity and antigenicity with a high score (0.8534). Additionally, the predicted vaccine contained no transmembrane helix regions, resulting in the easy expression of the vaccine 63. Therefore, the overall physical and chemical properties indicated that the vaccine should be considered heat-stable and eligible as a vaccine against cervical cancer.
Structural assessment of the chimeric vaccine was carried out by the assessment of the secondary and 3D of the vaccine structure. Analysis of the secondary structure demonstrated that the structure’s content comprised of α-helices, β-turns, extended strands and random coils. The best score generated by the 3D structure of the vaccine construct was selected and improved by a refinement tool so a more accurate template-based protein model nearer to the native state was obtained 64,65. To overcome one of the main problems faced in structural biology is how to recognize the errors in models of protein structures experimentally and theoretically 66. The ProSA tool was utilized for the prediction of the potential vaccine structural and modelling errors. In this report, the proposed vaccine revealed a Z-score of − 4.46 demonstrating the acceptable model of the vaccine.
Solubility is a critical protein structural property. It has important implications for therapeutics and diagnosis. The solubility of many proteins is low and resulted in heterologous overexpression of proteins and the formation of complex products 67,68. Multiple biochemical and functional investigations involve testing the solubility of the recombinant overexpressed protein in the E. coli host present 69,70. Protein sol server presented the vaccine as a soluble protein in comparison to E. coli proteins solubility. The predicted vaccine showed scaled solubility of 0.621 against 0.45 of the population’s E. coli solubility. Moreover, the disulfide bonds played a crucial role in protein structure stabilization and any strong disruption of these bonds is associated with loss of protein function and activity 71. Natural disulfide bonds significantly improve protein folding and stability 72,73. Also, the incorporation of new disulfide bonds into protein structures has been widely implemented to enhance protein stability, adjust functional properties and facilitate the investigation of protein dynamics 74,35. Furthermore, structural disulfide engineering reduces the number of potential conformations for a particular protein, lowering entropy and increasing thermostability 16. To introduce additional disulfide bonds into a refined model of the vaccine construct, the disulfide by design for disulfide engineering software was applied. The server predicted six disulfide bonds that stabilize the structure of the proposed vaccine. Moreover, the CLUSPRO docking server was utilized to study the TLR4-vaccine interaction. TLR4 is the primary stimuli receptor that triggers a proinflammatory response and also serves as an enhancer of the inflammatory response 75. The binding energy between the TLR4 chains and the vaccine demonstrated favourable interaction.
The most significant factor in the creation of recombinant proteins is the potential cloning of the designed vaccine in an appropriate E. coli expression vector. Before cloning into the pET30a (+) vector, the recommended vaccine was subjected to reverse transcription and modified for E. coli strain K. The CAI index was (0.954) and the GC content was 69.4 allowing for high levels of protein expression in bacteria. The vaccine construct was inserted within the multiple cloning site (MCS) of the vector molecule resulting in the successful cloning of the vaccine construct.
Conclusion
The vaccine constructs potentially induced cellular and humoral immune responses by combined B and T lymphocyte multi epitopes from L1 and L2 proteins using bioinformatics tools with no harmful effect to human. Thus the proposed vaccine would lead to be a suitable therapeutics protocol to combat HPV16. Nonetheless, the efficiency of vaccine must be approved with in vitro and vivo immunological analyzes.
Abbreviations
HPV: human papilloma virus; L1: major capsid protein; L2: minor capsid protein; ICC: invasive cervical cancer; IEDB: Immune Epitope Data Base web server; MHC-I: Major Histocompatibility Complex class I; MHC-II: Major Histocompatibility Complex class II; HLA: Human leucocyte antigen; ANN: Artificial Neural Network; NN-align: Neural Network align; pI: Isoelectric point; GRAVY: Grand average of hydropathicity; TLR4: Toll like Receptor 4; BLASTp: Basic local alignment search tool for protein; 3D structure: Three dimensional structure; PDBfile: Protein Data Bank file; QuerySol: Query solubility; PopAvrSol: Population average solubility; CAI: Codon adaptation index; JCAT: Java Codon Adaptation Tool; MCS: Multiple cloning site; OPSCC: oropharyngeal squamous cell carcinoma ; DbD2 : Disulfide by Design 2.0.
Conflict of Interest Statemen
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
Funding Sources
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
References
- Nwokolo NC, Barton SE. Sexually transmitted diseases of the vulva. Ridley’s The Vulva, Third Edition. 2009;6:44–70.
- De Sanjosé S, Diaz M, Castellsagué X, Clifford G, Bruni L, Muñoz N, et al. Worldwide prevalence and genotype distribution of cervical human papillomavirus DNA in women with normal cytology: A meta-analysis. Lancet Infect Dis. 2007;7:453–9.
- Arbyn M, de Sanjosé S, Saraiya M, Sideri M, Palefsky J, Lacey C, et al. EUROGIN 2011 roadmap on prevention and treatment of HPV-related disease. Int J Cancer. 2012;131:1969–82.
- Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907.
- de Sanjose S., Quint W.G., Alemany L., Geraets D.T., Klaustermeier J.E., Lloveras B., Tous S., Felix A., Bravo L.E., Shin H.R., et al. Human papillomavirus genotype attribution in invasive cervical cancer: A retrospective cross-sectional worldwide study. Lancet Oncol. 2010;11:1048–1056. doi: 10.1016/S1470-2045(10)70230-8.
- Giuliano A.R., Tortolero-Luna G., Ferrer E., Burchell A.N., de Sanjose S., Kjaer S.K., Munoz N., Schiffman M., Bosch F.X. Epidemiology of human papillomavirus infection in men, cancers other than cervical and benign conditions. Vaccine. 2008;26(Suppl. 10):K17–K28. doi: 10.1016/j.vaccine.2008.06.021.
- Sturgis EM, Cinciripini PM. Trends in head and neck cancer incidence in relation to smoking prevalence: an emerging epidemic of human papillomavirus-associated cancers? Cancer 2007; 110:1429-1435; PMID:17724670; http://dx.doi.org/10.1002/cncr.22963
- Chaturvedi AK, Engels EA, Anderson WF, Gillison ML. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J Clin Oncol 2008; 26:612-619; PMID:18235120; http://dx.doi.org/10.1200/JCO.2007.14.1713.
- Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology. 2010;401:70–79. doi: 10.1016/j.virol.2010.02.002.
- Burk RD, Harari A, Chen Z. Human papillomavirus genome variants. Virology. 2013;445:232–243. doi: 10.1016/j.virol.2013.07.018.
- Hung C-F, Wu TC, Monie A, Roden R. Antigen-specific immunotherapy of cervical and ovarian cancer. Immunological Reviews. 2008 Apr;222(1):43–69.
- de Villiers EM. Heterogeneity of the human papillomavirus group. J Virol. 1989;63:4898–4903.
- Pedersen C, Petaja T, Strauss G, et al. Immunization of early adolescent females with human papillomavirus type 16 and 18 L1 virus-like particle vaccine containing AS04 adjuvant. J Adolesc Health. 2007;40:564–71.
- Gall SA, Teixeira J, Wheeler CM, et al. Substantial impact on pre-cancerous lesions and HPV infections through 5.5 years in women vaccinated with the HPV-16/18 L1 VLP AS04 candidate vaccine. AACR Annual Meeting; Los Angeles, CA. 2007. Abstract Number: 4900.
- Almofti YA, Abd-elrahman KA, Eltilib EEM. Vaccinomic approach for novel multi epitopes vaccine against severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). BMC Immunology. 2021 Mar 25;22(1).
- Paul WE Fundamental Immunology, eds WE Paul, W Kluwer (Lippincott Williams & Wilkins, 7th Ed, New York, 2012).
- Kim Y, Ponomarenko J, Zhu Z, Tamang D, Wang P, Greenbaum J, Lundegaard C, Sette A, Lund O, Bourne PE, Nielsen M, Peters B. Immune epitope database analysis resource. Nucleic Acids Res. 2012;40(Web Server issue):W525–30. https://doi.org/10.1093/nar/gks438 Epub 2012 May 18. PMID: 22610854; PMCID: PMC3394288
- Sidney J, Assarsson E, Moore C, Ngo S, Pinilla C, et al. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 2008;4:2
- Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B. A Systematic Assessment of MHC Class II Peptide Binding Predictions and Evaluation of a Consensus Approach. Stormo G, editor. PLoS Computational Biology. 2008 Apr 4;4(4):e1000048.
- Dimitrov I, Bangov I, Flower DR, Doytchinova IA. AllerTOP v.2- a server for in silico prediction of allergens. J Mol Model. 2013;20:2278.
- Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R. Open source drug discovery consortium, Raghava GP in silico approach for predicting toxicity of peptides and proteins. PLoS One. 2013;8(9):e73957
- Tani K, Murphy WJ, Chertov O, Salcedo R, Koh CY, Utsunomiya I, Funakoshi S, Asai O, Herrmann SH, Wang JM, Kwak LW, Oppenheim JJ. Defensins act as potent adjuvants that promote cellular and humoral immune responses in mice to a lymphoma idiotype and carrier antigens. Int Immunol. 2000; 12(5):691–700 https://doi.org/10.1093/intimm/12.5.691.
- Ojha R, Pareek A, Pandey RK, Prusty D, Prajapati VK. Strategic development of a next-generation multi-epitope vaccine to prevent Nipah virus zoonotic infection. ACS Omega. 2019;4:13069–79.
- Shamriz S, Ofoghi H, Moazami N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput Biol Med. 2016;76:24–9. https://doi.org/10.1016/j.compbiomed.2016. 06.015 Epub 2016 Jun 17. PMID: 27393958.
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zheng Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
- Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schäffer AA, Yu Y-K. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005;272:5101–9.
- Rojas M, Restrepo-Jimenez P, Monsalve DM, Pacheco Y, Acosta-Ampudia Y, Ramírez-Santana C, Leung PSC, Ansari AA, Gershwin ME, Anaya JM. Molecular mimicry and autoimmunity. J Autoimmun. 2018;95:100–23.
- Combet C, Blanchet C, Geourjon C, Deléage G. NPS@: network protein sequence analysis. TIBS. 2000;25(3[291]):147–50.
- Källberg M., Wang H., Wang S., Peng J., Wang Z., Lu H., Xu J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012;7:1511–1522.
- Ko J, Park H, Heo L, Seok C. Galaxy WEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012;40(W1):W294–7.
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(Web Server issue):W407–10. https://doi.org/10.1093/nar/ gkm290.
- Hebditch M, Carballo-Amador MA, Charonis S, Curtis R, Warwicker J. ProteinSol: a web tool for predicting protein solubility from sequence. Bioinformatics. 2017;33(19):3098–100. https://doi.org/10.1093/bioinformatics/ btx345.
- Hasan M, Ghosh PP, Azim KF, Mukta S, Abir RA, Nahar J, Hasan Khan MM. Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus. Microb Pathog. 2019;130:19– 37. https://doi.org/10.1016/j.micpath.2019.02.023 Epub 2019 Feb 26.
- Niwa T, Ying BW, Saito K, Jin W, Takada S, Ueda T, Taguchi H. Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins. Proc Natl Acad Sci Unit States Am. 2009;106:4201–6.
- Craig DB, Dombkowski AA. Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinformatics. 2013;14:346. https:// doi.org/10.1186/1471-2105-14-346 PMID: 24289175.
- Vajda S, Yueh C, Beglov D, Bohnuud T, Mottarella SE, Xia B, Hall DR, Kozakov D. New additions to the ClusPro server motivated by CAPRI. Proteins: Structure, Function, and Bioinformatics. 2017 Mar; 85(3):435-444.
- Shey RA, Ghogomu SM, Esoh KK, Nebangwa ND, Shintouo CM, Nongley NF, Asa BF, Ngale FN, Vanhamme L, Souopgui J. In-silico design of a multiepitope vaccine candidate against onchocerciasis and related filarial diseases. Sci Rep. 2019;9(1):4409. https://doi.org/10.1038/s41598-019-40833-x
- Morla S, Makhija A, Kumar S. Synonymous codon usage pattern in glycoprotein gene of rabies virus. Gene. 2016;584:1–6.
- Pandey RK, Ojha R, Aathmanathan VS, Krishnan M, Prajapati VK. Immunoinformatics approaches to design a novel multi-epitope subunit vaccine against HIV infection. Vaccine. 2018;36:2262–72.
- Soria-Guerra RE, Nieto-Gomez R, Govea-Alonso DO, Rosales-Mendoza S. An overview of bioinformatics tools for epitope prediction: implications on vaccine development. J Biomed Inform. 2015 Feb;53:405-14. doi: 10.1016/j.jbi.2014.11.003. Epub 2014 Nov 10. PMID: 25464113.
- Khairkhah N, Namvar A, Kardani K, Bolhassani A. Prediction of cross-clade HIV-1 T-cell epitopes using immunoinformatics analysis. Proteins. 2018 Dec;86(12):1284-1293. doi: 10.1002/prot.25609. Epub 2018 Nov 1. PMID: 30260061.
- Li Y, Huang Y, Liang J, Xu Z, Shen Y, Zhang N, Liu Z, Zhao Y. Immune responses induced in HHD mice by multiepitope HIV vaccine based on cryptic epitope modification. Mol Biol Rep. 2013 Apr;40(4):2781-7. doi: 10.1007/s11033-012-2202-y. Epub 2013 Mar 1. PMID: 23456642.
- Oany AR, Emran AA, Jyoti TP. Design of an epitope-based peptide vaccine against spike protein of human coronavirus: an in silico approach. Drug Des Devel Ther. 2014 Aug 21;8:1139-49. doi: 10.2147/DDDT.S67861. PMID: 25187696; PMCID: PMC4149408.
- Chakraborty S, Chakravorty R, Ahmed M, Rahman A, Waise TM, Hassan F, Rahman M, Shamsuzzaman S. A computational approach for identification of epitopes in dengue virus envelope protein: a step towards designing a universal dengue vaccine targeting endemic regions. In Silico Biol. 2010;10(5-6):235-46. doi: 10.3233/ISB-2010-0435. PMID: 22430357.
- Hasan MA, Hossain M, Alam MJ. A computational assay to design an epitope-based Peptide vaccine against saint louis encephalitis virus. Bioinform Biol Insights. 2013 Nov 24;7:347-55. doi: 10.4137/BBI.S13402. PMID: 24324329; PMCID: PMC3855041.
- Gupta SK, Singh A, Srivastava M, Gupta SK, Akhoon BA. In silico DNA vaccine designing against human papillomavirus (HPV) causing cervical cancer. Vaccine. 2009 Dec 10;28(1):120-31. doi: 10.1016/j.vaccine.2009.09.095. Epub 2009 Sep 30. PMID: 19799841
- Ghorban Hosseini N, Tebianian M, Farhadi A, Hossein Khani A, Rahimi A, Mortazavi M, Hosseini SY, Taghizadeh M, Rezaei M, Mahdavi M. In Silico Analysis of L1/L2 Sequences of Human Papillomaviruses: Implication for Universal Vaccine Design. Viral Immunol. 2017 Apr;30(3):210-223. doi: 10.1089/vim.2016.0142. Epub 2017 Feb 16. PMID: 28388355.
- Singh KP, Verma N, Akhoon BA, Bhatt V, Gupta SK, Gupta SK, Smita S. Sequence-based approach for rapid identification of cross-clade CD8+ T-cell vaccine candidates from all high-risk HPV strains. 3 Biotech. 2016 Jun;6(1):39. doi: 10.1007/s13205-015-0352-z. Epub 2016 Jan 27. PMID: 28330110; PMCID: PMC4729761.
- Arshad M, Bhatti A, John P. Identification and in silico analysis of functional SNPs of human TAGAP protein: A comprehensive study. PLoS One. 2018 Jan 12;13(1):e0188143. doi: 10.1371/journal.pone.0188143. PMID: 29329296; PMCID: PMC5766082.
- Zepp F. Principles of vaccination. Vaccine Design: Springer; 2016. p. 57-84.
- Forster RJJop, methods t. Study designs for the nonclinical safety testing of new vaccine products. 2012;66(1):1-7.
- Konstantinou GN. T-Cell epitope prediction. Food Allergens: Springer; 2017. p. 211-22.
- Deng H, Yu S, Guo Y, Gu L, Wang G, Ren Z, et al. Development of a multivalent enterovirus subunit vaccine based on immunoinformatic design principles for the prevention of HFMD. 2020;38(20):3671-81.
- Bartlett BL, Pellicane AJ, Tyring SKJDt. Vaccine immunology. 2009;22(2):104-9.
- Chen X, Zaro JL, Shen W-C. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65(10):1357-69.
- Zhao HL, Yao XQ, Xue C, Wang Y, Xiong XH, Liu ZM. Increasing the homogeneity, stability and activity of human serum albumin and interferon-alpha2b fusion protein by linker engineering. Protein expression and purification. 2008;61(1):73-7.
- Amet N, Lee HF, Shen WC. Insertion of the designed helical linker led to increased expression of tf-based fusion proteins. Pharmaceutical research. 2009;26(3):523-8.
- Bai Y, Ann DK, Shen WC. Recombinant granulocyte colony-stimulating factor-transferrin fusion protein as an oral myelopoietic agent. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(20):7292-6.
- Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune TJPe. Design of the linkers which effectively separate domains of a bifunctional fusion protein. 2001;14(8):529-32.
- Feng Y, Jiang H, Qiu M, Liu L, Zou S, Li Y, Guo Q, Han N, Sun Y, Wang K, Lu L, Zhuang X, Zhang S, Chen S, Mo F. Multi-Epitope Vaccine Design Using an Immunoinformatic Approach for SARS-CoV-2. Pathogens. 2021 Jun 11;10(6):737. doi: 10.3390/pathogens10060737. PMID: 34208061; PMCID: PMC8230658.
- Kimple ME, Brill AL, Pasker RL. Overview of affinity tags for protein purification. Curr Protoc Protein Sci. 2013;73:9..1-9..23.
- Booth WT, Schlachter CR, Pote S, Ussin N, Mank NJ, Klapper V, et al. Impact of an N-terminal polyhistidine tag on protein thermal stability. 2018;3(1):760-8.
- Mohamed S, Almofti Y, Abd Elrahman KJCM. Exploring Crimean Congo Hemorrhagic Fever Virus Glycoprotein M to Predict Multi-Epitopes Based Peptide Vaccine Using Immunoinformatics Approach. 2021;10:122.
- Bhattacharya D. refineD: improved protein structure refinement using machine learning based restrained relaxation. Bioinformatics. 2019;35(18):3320-8.
- Pearce R, Zhang Y. Toward the solution of the protein structure prediction problem. Journal of Biological Chemistry. 2021;297(1):100870.
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research. 2007;35(suppl_2):W407-W10.
- Vihinen MJA, DMPK. Solubility of proteins. 2020;8(4):391-9.
- Smialowski P, Martin-Galiano AJ, Mikolajka A, Girschick T, Holak TA, Frishman D. Protein solubility: sequence based prediction and experimental verification. Bioinformatics. 2006;23(19):2536-42.
- Magnan CN, Randall A, Baldi P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics. 2009;25(17):2200-7.
- Vu TTT, Koo B-K, Song J-A, Chong S-H, Park CR, Nguyen MT, et al. Soluble overexpression and purification of bioactive human CCL2 in E. coli by maltose-binding protein. Molecular Biology Reports. 2015;42(3):651-63.
- Karimi M, Ignasiak MT, Chan B, Croft AK, Radom L, Schiesser CH, et al. Reactivity of disulfide bonds is markedly affected by structure and environment: implications for protein modification and stability. Scientific reports. 2016;6(1):38572.
- Zavodszky M, Chen CW, Huang JK, Zolkiewski M, Wen L, Krishnamoorthi RJPS. Disulfide bond effects on protein stability: Designed variants of Cucurbita maxima trypsin inhibitor‐V. 2001;10(1):149-60.
- Dombkowski AA, Sultana KZ, Craig DB. Protein disulfide engineering. FEBS letters. 2014;588(2):206-12.
- Zabetakis D, Olson MA, Anderson GP, Legler PM, Goldman ERJPO. Evaluation of disulfide bond position to enhance the thermal stability of a highly stable single domain antibody. 2014;9(12):e115405.
- Molteni M, Gemma S, Carlo R. The role of Toll-Like Receptor 4 in infectious and noninfectious inflammation. Mediators Inflamm Vol. 2016:Article ID 6978936, 9 https://doi.org/10.1155/2016/6978936.

This work is licensed under a Creative Commons Attribution 4.0 International License.



