

Manuscript accepted on : 18-10-2024
Published online on: 31-10-2024
Plagiarism Check: Yes
Reviewed by: Dr. Dinesh Kumar
Second Review by: Dr Durgesh Ranjan Kar
Final Approval by: Dr. Eugene A. Silow
Sivagami B1*, Dinesh MV1, Pavan Kumar V1, Chandrasekar R2 , Sanjeeva Kumar A2 and Satheesh Kumar G3
1Department of Pharmaceutical Analysis, Seven Hills College of Pharmacy, Tirupati, Andhra Pradesh, India.
2Department of Pharmacognosy, Seven Hills College of Pharmacy, Tirupati, Andhra Pradesh, India.
3Department of Pharmaceutical Chemistry, Seven Hills College of Pharmacy, Tirupati, Andhra Pradesh, India.
Corresponding Author E-mail:sivagamib_27@rediffmail.com
DOI : http://dx.doi.org/10.13005/bbra/3328

ABSTRACT: Ensitrelvir, is an oral SARS-CoV-2 3CL protease inhibitor that was approved in Japan to treat SARS-CoV-2 infections. This paper describes the AQbD approach and Box Behnken Design assisted development of a HPLC method and its validation for Ensitrelvir in bulk and dosage form. The three independent variables of the RP-HPLC method were flow rate, organic ratio in mobile phase and runtime and the responses retention time and tailing factor were taken as dependent variables. The study utilized a PLATSIL C18-EP column (4.6 x 250mm, 5µm) the chromatographic conditions were optimized using Acetonitrile: Triethylamine pH: 4 (60:40 mL) as the mobile phase, 1 mL/min as flow rate with a Rt of 9.609 min, at a λ max of 228 nm. The devised technique was found to be linear with a serial dilution of 10–50 μg/ml with (r2) of 0.991. The tailing factor (TF) and theoretical plates (N) were 1.28 and 4883 results indicated the system suitability test, respectively. The precision for Intraday and Interday were determined and % RSD was observed to be 1.6 and 0.9 %. The robustness values were below 2%. No other coeluting peaks were found with the Ensitrelvir peak, according to the chromatographic peak purity data. According to ICH specifications, the parameters for method validation were within the permissible range.
KEYWORDS:
Analytical Quality by Design; Box-Behnken Design; Ensitrelvir; Optimization; RP-HPLC
Download this article as:| Copy the following to cite this article: Sivagami B, Dinesh M. V, Kumar V. P, Chandrasekar R, Kumar A. S, Kumar G. S. Box-Behnken Design Assisted AQbD Approach for the Optimization and Quality Assessment of Ensitrelvir in Bulk and Dosage forms by RP-HPLC. Biotech Res Asia 2024;21(4). |
| Copy the following to cite this URL: Sivagami B, Dinesh M. V, Kumar V. P, Chandrasekar R, Kumar A. S, Kumar G. S. Box-Behnken Design Assisted AQbD Approach for the Optimization and Quality Assessment of Ensitrelvir in Bulk and Dosage forms by RP-HPLC. Biotech Res Asia 2024;21(4). Available from: https://bit.ly/48t2KO9 |
Introduction
In preclinical trial studies, the oral SARS-CoV-2 3C-like protease inhibitor ensitrelvir fumaric acid has exhibited antiviral activity which was active against variants of SARS-CoV-2, including subvariants of Omicron.1 Ensitrelvir has been approved since September 2023, it was approved for emergency use in Japan under Fast Track clinical trial conditions.2 The Registry of Clinical Trials Japan has conducted a clinical trial 2/3, randomized double-blind, placebo-controlled clinical trial.3 Treatment with ensitrelvir showed improvements in respiratory symptoms and in the multiple respiratory symptoms and slight feverishness was observed in phase 2b.4 In phases 2a and 2b it exhibited lower viral load compared to placebo. 5, 6
 |
Figure 1: Structure of EnsitrelvirClick here to view Figure |
The study was determined as per ICH Q8 for product development and ICH Q11 for manufacture and development of drug substance. 7, 8, 9 A well-understood product and process that reliably achieves its intended performance are the result of using QbD concepts. The information gathered throughout development can help construct a design space and choose appropriate process controls. The Analytical QbD (AQbD) refers to the use of these similar QbD concepts to the development of analytical procedures. 10
No analytical methods for quantifying ensitrelvir had previously been published, according to a thorough assessment of the literature. 11 12 This is the first method reported in literature for method development and validation for Ensitrelvir in dosage form or in its pure form. Developing and refining the Ensitrelvir HPLC technique in a solid dosage form through a QbD approach is the aim of this undertaking. 13, 14
Materials and Methods
Materials
Ensitrelvir was procured as complementary sample from MSN Lab Pvt. Ltd., Hyderabad. KH2PO4 was purchased from Finar chemical LTD, Acetonitrile, Methanol and Water for HPLC were obtained from Standard solutions Ltd, HCl, H2O2, NaOH procured from Merck Pvt. Ltd. Mumbai.
Instruments
A HPLC WATERS, software: Empowered, 2487 UV detector, was utilized for the study.
Chromatographic Conditions
The study utilized a PLATSIL C18-EP column (4.6 x 50 mm, 5µm) the chromatographic conditions were optimized using Acetonitrile: Triethylamine pH: 4 (60:40 mL) as the mobile phase, to adjust the mobile phase to pH 4, triethylamine (0.01%) was added. 1 mL/min as flow rate with a Rt of 9.609 min, at a λ max of 228 nm.
Preparation of Reference Standard Solution
By precisely dissolving 25 mg of Ensitrelvir into 25 ml VF, the standard stock solution was prepared. After diluting the stock solution, a sub-stock was prepared. Dissolving the solution was done with a sonicator. Using diluent to dilute the stock solution, 0.6 ml of the aforementioned solution was taken in a 10 ml VF.
HPLC Method Development by Analytical QbD
Selection of quality target product profile
Finding the variables that influence the QTPP parameters is a crucial task for the QTPP. For the suggested HPLC approach, QTPP was found to be the retention time and tailing factor. 13, 14
Determination of critical quality attributes
The CQAs selected for the study were runtime, flowrate and organic phase in mobile phase ratio considered to be independent variables and dependent variables (responses) were retention time and tailing factor.14
Factorial Design
The various interaction effects and quadratic influences of run time, organic phase in mobile phase ratio and flow rate on the retention time and tailing factor were investigated using a central composite statistical screening strategy. A 3-factor, flow rate, organic ratio in mobile phase and run time at three different levels were used in the design process using Design Expert software (Version 12.0). Three essential components were selected and optimized utilizing the central composite experimental design once the QTPP and CQAs were defined. 10 Because multivariable interactions between variables and process parameters have been explored, preliminary analysis was conducted to identify the components. 11 Table 1 lists the independent variables that were chosen, including run time, the organic phase in mobile phase and flow rate. The tailing factor and retention time were considered as the factors. 12
Analysing Experimental Findings and Choosing the Ideal Procedure Parameters
We evaluated these method variables using the CCD approach. The circumstances for the retention time and tailing factor have to be assessed in the first phase. This produced many chromatographic conditions for Ensitrelvir. Robust zones, wherein deliberate modifications to the procedure parameters have no effect on the quality, are found within the suggested acceptable limits. This technique ensures that validation testing can continue without concern for method failures in the future. Modifying the variable at different levels is necessary until the results fall within acceptable limits if the expected response from the modeling experiments is not attained. 15 The use of Design Expert tools optimized ideal chromatographic conditions.
Risk Assessment
The final optimized method was selected by taking into account factors like long-term functionality and dependability. The ICH Q8 and ICH Q9 guidelines’ QbD principles have an impact on the risk-based methodology used to assess the method’s robustness and ruggedness.7
Analytical Method Validation
Method validation is the process of confirming suitable analytical procedure for the purpose for which it is meant to be used. This can be confirmed using recorded evidence that provides a high level of certainty. The developed HPLC technique for Ensitrelvir estimation was evaluated in compliance with ICH Q2 (R1) requirements.9
Linearity
To examine the linearity of Ensitrelvir, five concentration ranges spanning from 10 to 50μg/ml were considered. The standard curve was plotted. The r2 value and the linear regression coefficient were calculated.
Precision
Repeatability was evaluated using measurements of six Ensitrelvir samples at 100μg/ml. Three distinct Ensitrelvir various concentrations were tested, once within the day at a 2-hour gap and on separate days in order to compute the intraday and interday precision. Less than two was the allowed range for the % RSD.
Accuracy
Recovery trials from commercial formulations at three distinct concentrations—50%, 100%, and 150% of standard addition—were used to evaluate the method’s accuracy. A recovery % was calculated for Esitrelvir. 98–102% of standard addition was the acceptable range for recovery % according to ICH recommendations.
LOD and LOQ
A drug’s LOD and LOQ refer to the lowest concentration at which a drug can be consistently detected and separated from the background and, respectively. To calculate the LOD and LOQ in accordance with ICH guidelines, the following formula was utilized.
σLOD=3.3×σ/SD
σLOQ=10×σ/SD
Robustness and Intermediate precision Studies
By changing minor adjustments to the procedure such as changing the pump flow rate and composition of mobile phase, the robustness of the methodology was evaluated. The analysis of the robustness studies required substituting an analyst for an extraneous influencing component. The allowed limit for the peak area’s estimated % RSD was below 2.
System Suitability Studies
Ensitrelvir was analysed six times in replicate to assess the system suitability. For standard solutions, the Rt, column efficiency, asymmetry factor and column efficiency were calculated.
Assay
Weigh out a powder to the exact quantity of 25 mg of Ensitrelvir and pour it into a 25 ml volumetric flask. Add diluent once the powder has completely dissolved, then sonicate for 15 minutes. After then, utilize the mobile phase to maintain the volume to desired level. The resulting solution can be filtered using 0.42 μ Whatman filter paper. HPLC was used to analyze the solution under the same chromatographic conditions as linearity. Three separate assays’ means were used for the calculation.
Results and Discussion
Initially, we tried a mobile phase ratio of ACN and water 50:50 v/v the retention time was too lengthy. The next mobile phase tried was ACN and water in the ratio 80:20 v/v, no peak was observed. Finally, we used Acetonitrile to Triethylamine in water at a ratio of 60:40 v/v. By modifying the pH of the buffer, the peak’s symmetry and shape were improved. The adjusted chromatographic conditions satisfied the specifications of the system suitability test. The ideal mobile phase was ACN to water 60:40 v/ v, with pH 4 adjusted with 0.01% triethylamine. Additional parameter optimization within the design space was conducted using the central composite design.
HPLC method development by QbD approach 14
Quality Target Product Profile
Retention time, theoretical plates, and peak asymmetry were the QTPP used in order to optimize the chromatographic conditions of HPLC.
Factorial Design 17
The Independent Variables (factors) and Dependent Variables (Responses) are shown in Table 1. Analysis of variance (ANOVA) test results are displayed in Table 2. Represents variables and their minimum and maximum levels are shown in Table 3, Represents Regression analysis for different responses and Fit Summary Table 4. Figures 2-6 represents the 2D contour plots for Retention time of Ensitrelvir, Figure 3 illustrates the 3D response surface plots for Retention time of Ensitrelvir, Figure 4 shows the 3D Surface for Tailing Factor of Ensitrelvir, Figure 5 illustrates the 2D contour plots for Tailing factor of Ensitrelvir, Figure 6 shows the Overlay plot for Ensitrelvir.
Table 1: Independent Variables (factors) and Dependent Variables (Responses)
| Std | Run | Factor 1 | Factor 2 | Factor 3 | Response1 | Response2 |
| A: Flow rate | B: Organic ratio in MP | C: run time | Retentiontime mins | tailingfactor | ||
| 6 | 1 | 1.00 | 55.00 | 14.00 | 9.5 | 1.27 |
| 4 | 2 | 1.00 | 60.00 | 14.50 | 9.6 | 1.28 |
| 12 | 3 | 0.90 | 60.00 | 15.00 | 9.4 | 1.6 |
| 10 | 4 | 0.90 | 60.00 | 14.00 | 9.2 | 1.6 |
| 15 | 5 | 0.90 | 55.00 | 14.50 | 8.5 | 1.6 |
| 2 | 6 | 1.00 | 50.00 | 14.50 | 8.2 | 1.6 |
| 1 | 7 | 0.80 | 50.00 | 14.50 | 8.6 | 1.6 |
| 9 | 8 | 0.90 | 50.00 | 14.00 | 8.6 | 1.6 |
| 17 | 9 | 0.90 | 55.00 | 14.50 | 8.5 | 1.6 |
| 3 | 10 | 0.80 | 60.00 | 14.50 | 8.3 | 1.6 |
| 7 | 11 | 0.80 | 55.00 | 15.00 | 8.7 | 1.6 |
| 16 | 12 | 0.90 | 55.00 | 14.50 | 8.5 | 1.6 |
| 5 | 13 | 0.80 | 55.00 | 14.00 | 8.7 | 1.6 |
| 13 | 14 | 0.90 | 55.00 | 14.50 | 8.5 | 1.6 |
| 8 | 15 | 1.00 | 55.00 | 15.00 | 8.3 | 1.6 |
| 14 | 16 | 0.90 | 55.00 | 14.50 | 8.5 | 1.6 |
| 11 | 17 | 0.90 | 50.00 | 15.00 | 8.6 | 1.6 |
Table 2: Analysis of variance (ANOVA) test results
| Source | Sum of Squares | df | mean Square | F-Value | p-value Prob > F | |
| Mean vs Total | 1291.96 | 1 | 1291.96 | Suggested | ||
| Linear vs Mean | 1.12 | 3 | 0.37 | 2.59 | 0.0972 | |
| 2FI vs Linear | 1.09 | 3 | 0.36 | 4.70 | 0.0269 | |
| Quadratic vs 2FI | 0.49 | 3 | 0.16 | 3.95 | 0.0611 | Suggested |
| Cubic vs Quadratic | 0.29 | 3 | 0.096 | 6.366E+007 | < 0.0001 | Aliased |
| Residual | 0.000 | 4 | 0.000 | |||
| Total | 1294.94 | 17 | 76.17 |
Table 3: Represents variables and their minimum and maximum levels
| Factor | Name | Type | Low Actual | High Actual | Low Coded | High Coded | Mean | Std. Dev. |
| A | Flow rate | Numeric | 0.80 | 1.00 | -1.000 | 1.000 | 0.900 | 0.069 |
| B | Organic mobile phase | Numeric | 50.00 | 60.00 | -1.000 | 1.000 | 55.000 | 3.430 |
| C | runtime | Numeric | 14.00 | 15.00 | -1.000 | 1.000 | 14.500 | 0.343 |
Table 4: Regression analysis for different responses and fit summary
| Response | Name | Units | Obs | Analysis | Minimum | Maximum | Mean | Std. Dev. | Ratio | Trans | Model |
| Y1 | Rt | min | 17 | Polynomial | 8.20 | 9.60 | 8.72 | 0.42 | 1.17 | None | No model chosen |
| Y2 | tailing factor | 17 | Polynomial | 1.27 | 1.60 | 1.56 | 0.10 | 1.26 | None | 2FI |
 |
Figure 2: 2D contour plots for Retention time of EnsitrelvirClick here to view Figure |
 |
Figure 3: The 3D response surface plots for Retention time of EnsitrelvirClick here to view Figure |
 |
Figure 4: 3D Surface for Tailing Factor of EnsitrelvirClick here to view Figure |
 |
Figure 5: 2D contour plots for Tailing factor of EnsitrelvirClick here to view Figure |
 |
Figure 6: Overlay plot for EnsitrelvirClick here to view Figure |
Design Space
The 17-run quadric design model, the RSM, and the CCD were used. After comparing the flow rate, mobile phase ratio and run time to the two responses, retention time, and tailing factor, the findings of the suggested CCD experimental design were presented. The flow rate has impact on tailing factor, when flow rate increases tailing factor decreases, run time decreases tailing factor decreases. The impact on retention time, organic phase decreases retention time decreases, runtime decreases retention time decreases. The observed value for responses was computed using the HPLC peak for the designated organic phase ratio and buffer pH in order to get the percentage of prediction error. A comparison with the anticipated values followed subsequently.
Method Validation
System Suitability
The system suitability test was examined by taking number of characteristics, including the Rt, which was determined to be 9.609 mins, the number of theoretical plates, 4883, the peak asymmetry, 1.28, and the % RSD of six injections was 0.82. Table 5 represents the results of system suitability parameters. Figure 7 depicts blank Chromatogram, Figure 8 represents Standard Chromatogram, Figure 9 displays Sample Chromatogram.
 |
Figure 7: system suitability Chromatogram for EnsitrelvirClick here to view Figure |
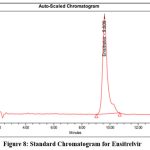 |
Figure 8: Standard Chromatogram for EnsitrelvirClick here to view Figure |
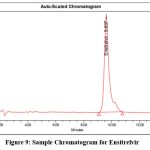 |
Figure 9: Sample Chromatogram for EnsitrelvirClick here to view Figure |
Table 5: Results of system suitability parameters
| S. No | Name | RT (min) |
Area (µV sec) |
Height (µV) |
USPtailing | USP plate count |
| 1 | Ensitrelvir | 9.609 | 308314 | 12899 | 1.28 | 4883 |
Linearity
Within the linear range of 10–50μg/ml, the regression line for Ensitrelvir that developed was linear, as shown in Figure 2 and Table 6. The calibration curve’s regression equation, y = 5038.6x + 3999.7, with a correlation coefficient of 0.9996, was often identified by plotting the graph of peak area vs concentration.
Table 6: Area of different concentration of Ensitrelvir
| S. No | Ensitrelvir | |
| Concentration (µg/ml) | Area | |
| 1 | 20 | 102771 |
| 2 | 40 | 205542 |
| 3 | 60 | 308314 |
| 4 | 80 | 411085 |
| 5 | 100 | 503856 |
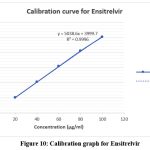 |
Figure 10: Calibration graph for EnsitrelvirClick here to view Figure |
Precision
Based on six injections of the same range, the % RSD for reliability for Ensitrelvir was observed to be 1.6 & 0.9. The developed method was considered to be precise if the % RSD value was below 2. The intraday and interday precisions values are displayed in Table 7.
Table 7: Results of Precision for Ensitreivir
| Injection | Area | Injection | Area |
| Injection-1 | 319439 | Injection-1 | 309439 |
| Injection-2 | 314607 | Injection-2 | 304607 |
| Injection-3 | 307106 | Injection-3 | 307106 |
| Injection-4 | 312764 | Injection-4 | 312764 |
| Injection-5 | 305953 | Injection-5 | 305953 |
| Injection-6 | 307962 | Injection-6 | 307962 |
| Average | 311305.2 | Average | 307971.8 |
| Standard Deviation | 5227.715 | Standard Deviation | 2872.803 |
| %RSD | 1.6 | %RSD | 0.9 |
Accuracy
Recovery study was used to ensure accuracy. Three different amounts of spiking were used to prepare sample solutions: 50%, 100%, and 150%. As per ICH Q2 (R1) requirements, the developed method was deemed accurate based on the recovery percentage of 98–102%. Table 8 displays the percentage recovery statistics that were acquired using the suggested HPLC procedure.
Table 8: Accuracy (recovery) data for Ensitrelvir
| %Concentration (at specification Level) | Area* | Amount Added (mg) | Amount Found (mg) | %Recovery | Mean Recovery |
| 50% | 99678.5 | 12.5 | 12.30 | 98.4 | 98.86 |
| 100% | 12988 | 25 | 24.92 | 99.68 | |
| 150% | 28451.9 | 37.5 | 36.95 | 98.5 |
Robustness studies
In order to assess robustness, a minor but deliberate modification of intrinsic technique characteristics including the pH and mobile phase flow rate was investigated. The ruggedness was examined as an unrelated contributing factor by a different analyst. A percentage RSD of less than 2 was obtained for the peak area due to variations in the mobile phase pH, the flow rate and the analyst. Ensitrelvir’s robustness results are shown in Tables 9 and 10.
Table 9: Results for variation in flow for Ensitrelvir
| S. No | Flow Rate (ml/min) | System Suitability Results | |
| USP Plate Count | USP Tailing | ||
| 1 | 0.8 | 4876 | 1.30 |
| 2 | 1 | 4883 | 1.28 |
| 3 | 1.2 | 4889 | 1.29 |
Table 10: Results for variation in mobile phase composition for Ensitrelvir
| S. No | Change in Organic Composition in the Mobile Phase | System Suitability Results | |
| USP Plate Count | USP Tailing | ||
| 1 | 10 % less (54ml) | 4880 | 1.30 |
| 2 | *Actual (60ml) | 4883 | 1.28 |
| 3 | 10 % more (66ml) | 4891 | 1.29 |
LOD and LOQ
The LOD and LOQ for Ensitrelvir were determined to be 2.96μg/ml and 9.97μg/ml. The results for LOD & LOQ are displayed in Table 11.
Table 11: Results of LOD & LOQ
| Drug name | Baseline noise(µV) | Signal obtained (µV) | S/N ratio | Conc. |
| Ensitrelvir | 83 | 246 | 2.96 | 0.1µg/ml |
| Ensitrelvir | 83 | 828 | 9.97 | 0.38µg/ml |
Assay
Using tablets for the experiment, the optimized chromatogram for Ensitrelvir revealed a resolved peak at retention time 9.607 min. Ensitrelvir’s label claim was found to have a drug content assay percentage of 100.5. The assay result showed that, in the presence of excipients included in tablet powder, the method was found to be more precise and specific.
Discussion
Using HPLC, an analytical QbD-driven method has been established for estimating ensitrelvir in solid dosage forms. The retention time and tailing factor served as the analytical target product profile for the Ensitrelvir HPLC investigation. Three components have been recognized as CQAs that impact the measurement of quality attributes (ATP): run time, flow rate, and organic phase in mobile phase. Using Design Expert Software Version 12.0, the CCD was applied for three variables at three separate levels. Important factors influencing the analytical target profile were found through a risk assessment analysis. .15, 16, 17 In order to retain control over variables related to column selection, instrument design, and injection volume, robustness study was assigned to govern several components of chromatographic separation, including flow rate and organic phase ratio of mobile phase. The study utilized a PLATSIL C18-EP column (4.6 x 250mm, 5µm) the chromatographic conditions were optimized using Acetonitrile: Triethylamine pH: 4 (60:40 mL) as the mobile phase, 1 mL/min as flow rate with a Rt of 9.609 min, at a λ max of 228 nm. The devised technique was observed to be with a linear range of 10–50μg/ml with (r2) of 0.991. The asymmetric factor (Tf) and theoretical plates (N) were 1.28 and 4883 results indicated the system suitability test. The precision for Intraday and interday were determined and found to be 1.6 and 0.9 % RSD. No other coeluting peaks were found with the Ensitrelvir peak, according to the chromatographic peak purity data. According to ICH specifications, the parameters for method validation were within the permissible range.
Conclusion
The current study offers a unique RP-HPLC method that was effectively developed and validated for estimation of Ensitrelvir. The use of AQbD with the Design expert Software significantly improved the method performance and robustness for successfully separating and estimating Ensitrelvir. The developed method was proven to be efficient for analysis of Ensitrelvir in bulk and tablet dosage forms. The technique minimizes the number of experimental runs and validation meet ICH Q2 R (1) requirements. The method could also be applied for the quantification of Ensitrelvir in dosage form and in its pure form.
Acknowledgement
Authors express their sincere gratitude to Seven Hills College of Pharmacy, Tirupati, for continuous motivation, support, and guidance for research activity and for providing all required facilities to accomplish the entitled work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Author Contributions
Chandrasekar R: Conceptualization, Methodology, Writing – Original Draft.
Sivagami B: Data Collection, Analysis, Writing – Review & Editing.
Pavan Kumar V: Visualization, Supervision, Project Administration.
Satheesh Kumar G: Funding Acquisition, Resources, Supervision
References
- Syed, Yahiya Y. “Ensitrelvir Fumaric Acid: First Approval.” Drugs 84,6 (2024): 721-728. doi:10.1007/s40265-024-02039-y.
CrossRef - Takazono T, Fujita S, Komeda T, Miyazawa S, Yoshida Y, Kitanishi Y, Kinoshita M, Kojima S, Shen H, Uehara T, Hosogaya N, Iwanaga N, Mukae H. Real-World Effectiveness of Ensitrelvir in Reducing Severe Outcomes in Outpatients at High Risk for COVID-19. Infect Dis Ther. 2024 Aug;13(8):1821-1833. doi: 10.1007/s40121-024-01010-4.
CrossRef - Yamato M, Kinoshita M, Miyazawa S, Seki M, Mizuno T, Sonoyama T. Ensitrelvir in patients with SARS-CoV-2: A retrospective chart review. J Infect Chemother. 2024;30(9):946-950. doi: 10.1016/j.jiac.2024.02.015.
CrossRef - Ogura E, Nakagawa N, Hayashi N, Tsukimura E, Takashima S. 537. Safety and Effectiveness of Ensitrelvir for the Treatment of COVID-19 in Japanese Clinical Practice: A Post-marketing Surveillance (Interim Analysis). Open Forum Infect Dis. 2023;10(Suppl 2): ofad500.606. doi:10.1093/ofid/ofad500.606.
CrossRef - Yotsuyanagi H, Ohmagari N, Doi Y, Yamato M, Bac NH, Cha BK, Imamura T, Sonoyama T, Ichihashi G, Sanaki T, Tsuge Y, Uehara T, Mukae H. Efficacy and Safety of 5-Day Oral Ensitrelvir for Patients With Mild to Moderate COVID-19: The SCORPIO-SR Randomized Clinical Trial. JAMA Netw Open. 2024 Feb 5;7(2):e2354991.
CrossRef - Lin, M., Zeng, X., Duan, Y. Molecular mechanism of ensitrelvir inhibiting SARS-CoV-2 main protease and its variants. Commun Biol2023; 6, 694. https://doi.org/10.1038/s42003-023-05071-y.
CrossRef - Mukae H, Yotsuyanagi H, Ohmagari N, Doi Y, Sakaguchi H, Sonoyama T, Ichihashi G, Sanaki T, Baba K, Tsuge Y, Uehara T. Efficacy and Safety of Ensitrelvir in Patients With Mild-to-Moderate Coronavirus Disease 2019: The Phase 2b Part of a Randomized, Placebo-Controlled, Phase 2/3 Study. Clin Infect Dis. 2023 Apr 17;76(8):1403-1411. doi: 10.1093/cid/ciac933.
CrossRef - The International Conference on Harmonization ICH Technical Requirements for Registration of Pharmaceuticals for Human Use on Pharmaceutical Development Q8(R2) (2009) https://database.ich.org/sites/default/files/Q8%28R2%29%20Guideline.pdf
- The International Conference on Harmonization ICH Technical Requirements for Registration of Pharmaceuticals for Human Use on Quality Risk Management Q9 (2005) https://database.ich.org/ sites/default/files/Q9%20Guideline.pdf
- The International Conference on Harmonization ICH Technical Requirements for Registration of Pharmaceuticals for Human Use on Pharmaceutical Quality System Q10 (2008) https://database.ich.org/sites/default/files/Q10%20Guideline.pdf
- Patel, K.Y., Dedania, Z.R., Dedania, R.R. QbD approach to HPLC method development and validation of ceftriaxone sodium. Futur J Pharm Sci2021; 7, 141. https://doi.org/10.1186/s43094-021-00286-4.
CrossRef - Peraman R, Bhadraya K, Reddy YP, Reddy CS, Lokesh T. Analytical Quality by Design Approach in RP-HPLC Method Development for the Assay of Etofenamate in Dosage Forms. Indian J Pharm Sci. 2015;77(6):751-757. doi:10.4103/0250-474x.174971
CrossRef - Urmi, K.F., Nawaz, M.S. & Islam, S.M.A. Analytical quality by design approach to RP-HPLC method development and validation for simultaneous estimation of esomeprazole and naproxen in modified-release dosage form. Futur J Pharm Sci2022; 8, 8. https://doi.org/10.1186/s43094-021-00396-z.
CrossRef - Park G, Kim MK, Go SH, Choi M, Jang YP. Analytical Quality by Design (AQbD) Approach to the Development of Analytical Procedures for Medicinal Plants. Plants (Basel). 2022;11(21):2960. doi:10.3390/plants11212960.
CrossRef - Tim Tome Nina Žigart Zdenko Časar Aleš Obreza. Organic Process Research & Development Process Res. Dev. 2019; 23, 9, 1784–1802. https://doi.org/10.1021/acs.oprd.9b00238.
CrossRef - Verch T, Campa C, Chéry CC, Analytical Quality by Design, Life Cycle Management, and Method Control. AAPS J. 2022;24(1):34. doi:10.1208/s12248-022-00685-2.
CrossRef - Nuli, M.V., Seemaladinne, R. & Tallam, A.K. Analytical quality by design (AQbD) based optimization of RP-UPLC method for determination of nivolumab and relatlimab in bulk and pharmaceutical dosage forms. Futur J Pharm Sci2024; 10, 86. https://doi.org/10.1186/s43094-024-00659-5.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.



