

Manuscript accepted on : August 25, 200
Published online on: 28-12-2009
Expression and Purification of Yeast Erf1 Mutant Proteins in Escherichia Coli
Fida Madayanti1, Prima Endang Susilowati1,3 Pingkan Aditiawati2 and Akhmaloka1*
1Biochemistry Research Group, Faculty of Mathematics and Natural Sciences, Institut Teknologi Bandung, Jln Ganesha 10, Bandung Indonesia.
2School of Life Science and Technology, Institut Teknologi Bandung, Jln Ganesha 10, Bandung Indonesia.
3Departement of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Haluoleo, Kendari Indonesia.
ABSTRACT: Translation termination in eukaryotes is mediated by two release factors, eRF1 and eRF3, which interact to form a heterodimer that mediates termination at all three stop codons. Detailed mechanism of the interaction between the two proteins, however, was still unclear yet. Previous studies indicated that threonin-295 on the third domain of eRF1, involved in its interaction to eRF3. In other to further characterize the role of threonine at position 295 on its interaction, two SUP45 mutants, namely sup45-T295A and sup45-T295S, were constructed and expressed in Escherichia coli. The mutations were successfully performed by PCR megaprimer and confirmed by sequence analysis. The mutant genes were over expressed in Escherichia coli BL21(DE3) under the promotor of T7 using pUKC630 vector. The mutant proteins namely, eRF1-T295A and eRF1-T295S, were expressed over 19% and 18% of total protein, respectively. The proteins were successfully purified by one step purification through Immobilized Metal Affinity Chromatography (IMAC).
KEYWORDS: ERF1; sup45 mutants; over-expression; IMAC purification
Download this article as:| Copy the following to cite this article: Madayanti. F, Susilowati. P. E, Aditiawati P, Akhmaloka. Expression and Purification of Yeast Erf1 Mutant Proteins in Escherichia Coli. Biosci Biotechnol Res Asia 2009;6(2) |
| Copy the following to cite this URL: Madayanti. F, Susilowati. P. E, Aditiawati P, Akhmaloka. Expression and Purification of Yeast Erf1 Mutant Proteins in Escherichia Coli. Biosci Biotechnol Res Asia 2009;6(2). Available from: https://www.biotech-asia.org/?p=8621 |
Introduction
Protein biosynthesis terminates when one of the three stop codons enters the aminoacyl site (A-site) and signals polypeptide chain release from the peptidyl-tRNA located in the ribosomal P-site 1. The process is facilitated by two general groups of accessory proteins: a class I release factor, codon-specific RFs (RF1 and RF2 in prokaryotes; eRF1 in eukaryotes), and a class II release factor, codon-non specific RFs (RF3 in prokaryotes and eRF3 in eukaryotes) that binds guanine nucleotides-binding proteins possessing GTPase activity 2, 3. Although the basic biological action of class-1 RFs is similar between prokaryotes and eukaryotes, they exhibit distinct structural and functional features. The release factor 1 (eRF1) recognizes all three stop codons and promotes the activation of the peptidyl transferase center, leading to the delivery of the nascent polypeptide 4. Moreover eukaryotes release factor 3 (eRF3) has GTPase activity that enhances the activity of eRF1 5.
The yeast eRF1 protein (the product of the essential SUP45 gene) does not share significant sequence homology with its prokaryotic counterparts 6. eRF1 is comprised of three distinct domains 7. Domain 1 includes the conserved amino acid motifs YxCxxxF (yeast amino acid residues 122 to 128) and TASNIKS (yeast amino acid residues 55 to 61), which have been implicated in stop codon binding. Domain 2 contains the conserved GGQ motif (yeast amino acid residues 180 to 182), which is responsible for the peptidyl transferase hydrolytic activity 8. Finally, domain 3 of eRF1 was suggested to be mediated its association with eRF3 7. Progressive deletion of the C-terminal 6-19 amino acids in Saccharomyces cerevisiae 9 and 17 amino acids of Schizusacharomyces pombe 3 resulted in a corresponding loss of eRF3 binding. In any case, the core of eRF3-binding region identified for Homo sapiens eRF1 (by these yeast two-hybrid deletion analysis), showed that two regions in each release factor were critical for mutual binding, position at 281-305 and 411-415 (GILRY) of eRF1 and position at 478-530 and 628-637 of eRF3 10, 11. Although deletion of residues within the third domain of eRF1 resulted in the loss of eRF3 interaction, however detail mechanism of translation termination, including the role of eRF1, were still unclear yet.
Computer modeling analysis showed that tyrosine at position of 410 (Y410) and threonine at position of 295 (T295) of eRF1 exposed to the surface of molecules and predicted to be involved in its interaction to eRF3 11, 12. Mutation on tyrosine to serine at position of 410 in eRF1 has been reported to decrease the binding affinity of the protein to eRF3 protein11. In addition, mutation of threonine at position 295 increased the suppression of termination codons 13. In order to further probe the role of T295 in yeast eRF1 protein, here we reported the construction and expression of sup45 mutants in E. coli. The protein mutants were successfully purified by IMAC system.
Material and Methods
Plasmids, microbial strains, and growth conditions
Plasmids used in this study were pUKC1901 as source of SUP45 gene and pUKC630 as expression vector in E. coli (Table 1). E. coli strains used in the study were DH5a [supE44 lacU169 (Q80lacZ M15) hsdR17 recA1 endA1 gyrA96 thi-1 rei A1] used for cloning experiments and BL21(DE3) [HsdS gal (lcIts857 ind1 Sam7 nin5 lacUV5-T7 gene 1)] used as host for expression of the proteins. E. coli cultures were grown in Luria Bertani medium (LB) (1% (w/v) bacto tryptone, 0.5% (w/v) yeast extract, and 0.5% (w/v) NaCl) supplemented with relevant antibiotics for selection.
Table 1: Plasmid used in this study.
| Plasmid | Description | Source |
| pUKC1901 | Shuttle vector in E. coli and S. cerevisiae. Carrying the SUP45 gene | University of Kent at Canterbury England |
| pUKC630 | Multi-cloning-site expression vector with a hexa-histidine sequence under the control of T7 RNA polymerase promotor. Carrying the SUP45 gene | University of Kent at Canterbury England |
| pUKC630-T295A | pUKC630 that SUP45 gene was replaced by sup45-T295A | This study |
| pUKC630-T295S | pUKC630 that SUP45 gene was replaced by sup45-T295S | This study |
PCR Megaprimer
The mutagenesis procedure was performed according to PCR-bases “Megaprimer” method 14, 15. Two steps of PCR were performed to construct sup45 mutant genes. The first PCR amplified DNA fragments which were used as megaprimers. The PCR was performed using PT295A or PT295S and PRSUP45 primers (Table 2). The second PCR amplified the whole SUP45 gene fragment using PFSUP45 and the mega primer. The first PCRs were carried out in 25 mL reaction mixtures containing 50 ng of pUKC1901 DNA, 10 ng forward primers (PT295A or PT295S) and 10 ng reverse primer (PRSUP45), 0.2mM each of four deoxyribonucleoside triphosphates, 1x of Pfu DNA polymerase reaction buffer and 1.25 U of Pfu DNA polymerase (Promega). The denaturation was carried out at 940C (4 min) for the first cycle, and at 940C (90s) for the next 25 cycles. The annealing temperature was carried out at 39oC (120s) and the elongation at 720C for 90s. The second PCRs were performed similar to that the first one, instead of the primers used and the PCR mixture. The forward primer was PFSUP45 and the reverse primer was the product of the first PCR (megaprimer). In the end of first PCR, the mixture was added with 10 ng forward primer (PFSUP45). The PCR process was continue with the same cycle as the firs PCR.
Table 2: The primers used in this study. Underlined showed the mutation created.
| Name | Sequences |
| PT295A
PT295S PFSUP45 PRSUP45 |
CCAGGACGCAGGTAAATTCTG
CCAGGACTCAGGTAAATTCTG GAAGGTCAAGAAGTTGGTC GCGAAAGGGGGATGTG |
Cloning of sup45 mutants
The full-length of sup45 genes from the result of second PCR were cloned into pUKC630 through BamHI and EcoRV restriction sites. The PCR fragments were cut by BamHI and EcoRV restriction enzymes. The fragments were inserted to pUKC630 following digestion with the same enzymes. The ligation was performed at 12oC for 18 h. The ligation mixture was used to transform E. coli DH5a. The recombinant plasmids were isolated by using QIAprep Spin Miniprep Kit (Qiagen). The recombinant plasmids were verified by restriction digest and DNA sequencing analysis. The appropriate clones were used for expression of the eRF1 mutants.
Expression of yeast eRF1
The recombinant plasmid, pUKC630 and pUKC630-sup45 mutants containing His-tag gene at the upstream region of SUP45 or sup45 coding sequences were introduced into E. coli BL2(DE3) as host strain for eRF1 expression system. For expression of the gene, the transformants were grown aerobically at 370C in LB containing ampicillin until the cells density reached 2 X 108 cell/mL or OD600 at around 0.6. The cultures were induced by addition of 0.1 mM IPTG and incubated at 250C for 4 h with aeration. The cultures were then centrifuged at 5000 g and the pellets were re-suspended in the buffer (50mM Tris-HCl pH 7.4; 200 mM NaCl). Lysis cells were performed by sonication. The samples were centrifuged at 10000 g and then analyzed by sodium dodecyl sulfate polyacrylamid gel electrophoresis (SDS-PAGE).
Protein purification and SDS-PAGE
Histag-eRF1 fusion proteins were purified under denatured conditions using Ni-NTA column (Qiagen, GmbH, Germany). Soluble proteins were load onto Ni-NTA column equilibrated with A (binding) buffer (300 mM NaCl; 50 mM sodium phosphate buffer; 10 mM imidazole; pH 8.0). The column was washed with B (washing) buffer (300 mM NaCl; 50 mM sodium phosphate buffer; 20 mM imidazole; pH 8.0), and Histag-eRF1 fusion protein was eluted with C (eluting) buffer (300 mM NaCl; 50 mM sodium phosphate buffer; 100-200 mM imidazole; pH 8.0).
The purified proteins were subjected to SDS-PAGE. The SDS-PAGE was performed with a 12.5% (w/v) acrylamide gel, and the proteins were stained with commassie brilliant blue G-250 16 .
Result and Discussion
Construction of sup45 mutants
The previous study showed that two regions in eRF1 were identified as critical regions for mutual binding of the protein to eRF3 7, 17, 9, 11 . In human eRF1 the regions lied at the position of 281 – 305 and 411 – 415 7. Computer analysis on the structure of yeast eRF1 showed that threonine at position 295 and tyrosine at 410 were found on a bend of the turn region. These two amino acid residues were proposed to be involved on the interaction of eRF1 to eRF3 proteins 18.
Two sup45 mutant genes, namely sup45-T295A and sup45-T295S encoding for eRF1-T295A and eRF1-T295S, were constructed based on PCR megaprimer method 14, 15. Two steps of PCR were carried out to amplify the whole coding region of sup45 mutants (Fig 1). PCR megaprimer has some advantages compared to that the overlap extension 19, 15. This method only used two steps of PCR while the overlap extension needs three steps. As consequence, the overlap extension needs more primers than the megaprimer method 20, 21.
 |
Figure 1. Strategy used for construction of sup45 mutant genes. A: primer PFSUP45, B: primer PRSUP45, M: Primer PT295A or PT295S. PCR I, amplifying DNA fragment using PT295A or PT295S and PRSUP45 primers, which were used as mega primers. PCR II, amplified the whole SUP45 gene fragment using PFSUP45 and the mega primer.
|
Construction of each mutant using megaprimer method in this experiment required 3 primers (Table 2). The first PCR was performed using PT295A or PT295S and PRSUP45 primers resulting amplicon at around 850 bp (Fig 2). The second PCR was carried out using PFSUP45 and the megaprimer (amplicon from the first PCR), resulting the fragment DNA at around 1800 bp in size (Fig 2). The successful of the first PCR to get the amplicon is determined by the primers used, while for the second PCR, the length of the megaprimer was important to get good amplification 22. The ideal size of megaprimer was at around 200 – 500 bp, however in this experiment the size much longer than that the ideal (850 bp). The successful to get the amplicons from the second PCR was probably due to on the manipulation of period and temperature of annealing process. The mutation on the sup45 mutant genes were confirmed by nucleotide sequence analysis (Fig 3).
 |
Figure 2: Electrophoregram of PCR product. Lane 1, pUC19 digest by HinfI as marker DNA ; lane 2, the product of first PCR for sup45-T295A (850 pb); Lane 3, the product of first PCR for sup45-T295S (850 pb); lane 4, the product of second PCR for sup45-T295A (1800 pb); lane 5, the product of second PCR for sup45-T295S (1800 pb); lane 6, l DNA digest by HindIII as marker DNA.
|
 |
Figure 3: Sequence comparison. (A) Comparison between SUP45 and sup45-T295A gene, meanwhile, (B) Comparison between SUP45 and sup45 – T295S gene. Underline showed the nucleotide changes.
|
Expression and purification of eRF1
The whole coding region of sup45-T295A and sup45-T295S were successfully cloned into plasmid pUKC630 through BamHI and EcoRV restriction sites resulting on pUKC630-T295A and pUKC630-T295S respectively (Fig 4). In this plasmid the gene was controlled under the promoter of bacteriophage T7. The plasmid were used to transform E. coli BL21(DE3) which was suitable for the expression of the plasmid since this strain carrying T7 RNA Polymerase from bacteriphage DE3 23(Studier and Moffat, 1986). The gene under T7 promotor remaining transcriptionally silent until the expression a chromosomal copy of T7 RNA Polymerase was induced. The expression of eRF1 mutants were carried out under the induction of IPTG when the culture cells reached on logarithmic phase. The results showed that the proteins were over-expressed up to 19% of the total protein (Fig 5), following densitometric measurement on the gel (data not shown).
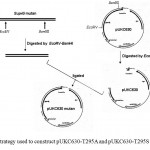 |
Figure 4: Strategy used to construct pUKC630-T295A and pUKC630-T295S.
|
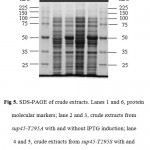 |
Figure 5: SDS-PAGE of crude extracts. Lanes 1 and 6, protein molecular markers; lane 2 and 3, crude extracts from sup45-T295A with and without IPTG induction; lane 4 and 5, crude extracts from sup45-T295S with and without IPTG induction.
|
Purification of the eRF1 mutants were performed by IMAC system since the eRF1 expressed under pUKC630 containing the His tag (6 histidine residues) in the N-terminal of the protein. The purification using IMAC system was based on the specific interaction between transitional metal ion (Co2+ or Ni2+) with a donor electron from the amino acid residues 24, 25. In the case of eRF1 mutants, the histidine was used as a donor electron and Ni2+ was the acceptor. The soluble crude extract of eRF1 mutants were directed subjected to Ni-NTA affinity chromatography. The eRF1 proteins were eluted by the addition of 100 mM immidazoles. SDS-PAGE analysis showed that the single band of 49 kDa of protein was present in all fractions (Fig 6).
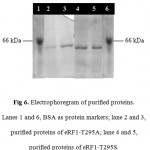 |
Figure 6: Electrophoregram of purified proteins. Lanes 1 and 6, BSA as protein markers; lane 2 and 3, purified proteins of eRF1–T295A; lane 4 and 5, purified proteins of eRF1–T295S.
|
In vivo functional studies of eRF1 were carried out through analysis of nonsense codon suppression 12, 11. Mutation of Thr at position 295 to Ala or Ser increased stop codons suppression. The high stop codon suppression might be due to the slight modification of the structure of the C-terminal motif 13. However, there is no information concerning in vitro studies of the proteins. The success of expression and purification of the proteins allow the in vitro study of the proteins to be carried out.
Conclusion
Two SUP45 mutants, namely sup45-T295A and sup45-T295S, were successfully constructed using PCR megaprimer without fragment purification. The mutations have been confirmed by nucleotide sequence analysis. The gene were expressed in E. coli BL21(DE3) encoded 49 kDa protein in size under the promoter of T7. Immobilized Metal Affinity Chromatography was successfully used to purify the proteins.
Acknowledgment
We would like to thanks to Prof M.F. Tuite for the gift of plasmid and strains. This research was supported by Fundamental Research Grant, Department of National Education, Republic of Indonesia to AKH and BPPS Scholarship to PES.
References
- Inge-Vechtomov S., Zhouravleva, G., and Philippe, M. Eukaryotic Release Factor (eRFs) History (Review), Biology of the Cell, 2003; 95: 95-209.Stansfield I., Jones, K.M., Kushnirov, V.V., Dagkesamanskaya, A.R., Poznyakovski, A.I., Pauskhin, S.V., Nierras, C.R., Cox, B.S., Teravanesyan, M.D., and Tuite, M.F. The Products of the SUP45 (eRF1) and SUP35 Genes Interact to Mediate Translation Termination in Saccharomyces cerevisiae. EMBO J., 1995; 14: 4365-4373.
- Ito K., Uno, M., and Nakamura, Y. Single Amino Acid Substitution in Prokaryotic Polypeptide Release Factor 2 Permits it to Terminate Translation at all Three Termination Codons, Proc Natl Acad Sci USA, 1998; 95: 8165-8169.
- Frolova L.Y., Le, G.X., Rasmussen, Y., Cheperegin, S., Drugeon, G., Kress, M., Arman, Haenni, A.L., Celis, J.E., Philippe, M. , and Kisselev, L. A Highly Conserved Eukaryotic Protein Family Possessing Properties of Polypeptide Chain Release Factor, Nature, 1994; 372: 701-703.
- Zhouravleva G., Frolova, L.Y., Le, G.X., Le Guellec, R., Inge-Vechtomov, S., Kisselev, L., and Philippe, M. Termination of Translation in Eukaryotes in Governed by Two Interacting Polypeptide Chain Release Factor, eRF1 and eRF3, EMBO J., 1995; 14: 4065-4072.
- Stansfield I., Eurwilaichitr, L., Akhmaloka, and Tuite, M.F. Depletion in the Levels of the Release Factor eRF1 Cause a Reduction in the Efficiency of Translation Termination in Yeast, Mol. Microbiol, 1996; 20: 1135-1143.
- Frolova L. Y., T. I. Merkulova, T.I., and Kisselev, L.L. Translation Termination in Eukaryotes: Polypeptide Release Factor eRF1 is Composed of Functionally and Structurally Distinct Domains, RNA, 2000; 6: 381-390.
- Frolova L. Y., Tsivkovskii, R.Y., Sivolobova, G.F., Oparina, N.Y., Serpinsky, O.I., Blinov, V.M., Tatkov, S.I., and Kisselev, L.L. Mutations in the Highly Conserved GGQ Motif of Class 1 Polypeptide Release Factor Abolish Ability of Human eRF1 to Trigger Peptidyl-tRNA Hydrolysis, RNA, 1999; 5: 1014-1020.
- Eurwilaithtr L., Graves, F.M., Stansfield, I., and Tuite, M.F.The C Terminus of eRF1 Define a Functionally Important Domain for Translation Termination in Saccharomyces cerevisiae, Mol. Microbial, 1999; 32: 485-496.
- Merkulova T. I., Frolova, L.Y., Lazar, M., Camonis, J., and Kisselev, L.L. C-Terminal Domains of Human Translation Termination Factors eRF1 and eRF3 Mediated their In Vivo Interaction, FEBS Letter, 1999; 443: 41-47.
- Akhmaloka, Susilowati, P.E., Subandi, and Madayanti, F. Mutation at Tyrosine in AMRLY (GIRLY like) motif of yeast eRF1 on nonsense codons suppression and binding affinity to eRF3. Int J Biol Sci, 2008; 4: 87-95.
- Susilowati P.E., Aditiawati, P. Madayanti, F., and Akhmaloka. Mutant of Y410A on AMRLY Motif of Yeast eRF1 Enhance Suppression of Nonsense Codons in Saccharomyces cerevisiae, IJIB; 2008; 3: 84-91.
- Susilowati P.E., Aditiawati, P., Madayanti, F., and Akhmaloka. The Effect of Mutation at Thr 295 of Saccharomyces cerevisiae eRF1 on Suppression of Nonsense Codons and eRF1 Structure, Microbiology Indonesia, 2008; 2: 11-16.
- Ambarsari L., Proamono, H. Madayanti, F., Moeis, M.R., and Akhmaloka. Konstruksi dan Overekspresi Gen Penyandi DNA Polimerase I Mutan dari Bakteri Termofil Isolat Lokal, Hayati, 2004; 11: 115-120.
- Tyagi R., R. Lai, R., and Duggleby, R.G. A New Approach to ‘Megaprimer’ Polymerase Chain Reaction Mutagenesis without an Intermediated Gel Purification Step, BMC Biotechnology, 2004; 4: 2.
- Sambrook J., Fritsch, E.F., and Maniatis, T. Molecular Cloning: a Laboratory Manual, Cold Spring Harbor, Cold Spring Harbor Press, 1989.
- Ebihara K., and Nakamura, Y. C-Terminal Interaction of Translational Release Factor eRF1 and eRF3 of Fission Yeast: G-Domain Uncoupled Binding and the Role of Conserved Amino Acid, RNA, 1999; 5: 739-750.
- Subandi, Pengaruh Mutasi Y410S eRF1 pada Interaksi eRF1-eRF3 Saccharomyces cerevisiae [Disertasi Doktor], Institut Teknologi Bandung, Bandung, 2002.
- Barik S. Mutagenesis and Gene Fusion by Megaprimer PCR, in White, B. A., (eds), PCR Cloning Protocols, New Jersey: Humana Pr., 1997; 173-182.
- Newton C.R., and Graham, A. PCR: the Introduction toor Biotechniques, 2nd ed., Oxford: Bios Scientific, 1994.
- Horton R.M. In Vitro Recombinant and Mutagenesis of DNA, in White, B. A., (eds), PCR Cloning Protocols, New Jersey: Humana Pr., 1997, 173-182.
- Pons J., Planas, A., Juncosa, M., and Querol, E. PCR Site-Directed Mutagenesis using Pyrococus, sp. GB-D-Polymerase Coupled to a Rapid Screening procedure, in White, B. A., (eds), 1997, PCR Cloning Protocols, New Jersey: Humana Pr., 1997, 173-182 .
- Studier F.M., and Moffat, B.A. Use of Bacterio Phage T7 RNA Polymerase to Direct Selective High Level Expression of Cloned Genes, J. Mol. Biol, 1986; 189: 113-130.
- Hochuli E., Bannwarth, W., Dobeli, H., Gentz, R., and Stuber, D. Genetic Aproach to Facilitate Purification of Recombinant Proteins with a Novel Metal Chelate Adsorbant, Bio/Technology, 1998; 6: 1321-1325.
- Van Reth T., Dreze, P.L., Szpirer, J., Szpirer, C., and Gabant, P. Positive Selection Vectors to Generate Fused Genes for the Expression of His-TaggedProteins, BioTechniques, 1998; 25: 898-904

This work is licensed under a Creative Commons Attribution 4.0 International License.



