

Manuscript accepted on : February 19, 2011
Published online on: 28-06-2011
M. Kalantari, M. Motallebi* and M. R. Zamani
National Institute of Genetic Engineering and Biotechnology (NIGEB), Tehran, I.R. of Iran
ABSTRACT: Polygalacturonase-inhibiting proteins (PGIPs) selectively inhibit the polygalacturonases (PGs) secreted by the invading plant pathogenic fungi. The PGIPs display the differential inhibiting towards the PGs from different fungi, also towards the different isoforms of the PGs originating from the specific pathogen. In this study the extracts from the Phaseolus vulgaris (cv. Emerson) hypocotyle inhibited the crude protein containing the cell wall degrading polygalacturonase activity of Rhizoctonia solani AG2-2, the causal agent of the root and stem rot on the sugar beet. This inhibitory activity has not yet been linked conclusively to the activity of the pgip2 gene product. In this study, after isolation and cloning of the pgip2 gene from bean, we used a transgenic over-expression approach to show that the PGIP encoded by the pgip2 gene is active against the PGs of R. solani. To assess the effectiveness of these proteins in protecting the sugar beet from the fungal pathogens, a number of the transgenic sugar beet lines expressing a bean pgip2 were produced. Independent transgenic lines were characterized by PCR, southern dot blot, agarose diffusion assay and ELISA. The presented data confirm the antifungal nature of the pgip2 gene.
KEYWORDS: pgip2; sugar beet; Rhizoctonia solani; PGIP; polygalacturonase
Download this article as:| Copy the following to cite this article: Kalantari. M, Motallebi. M, Zamani. M. R. Bean Polygalacturonase-Inhibiting Protein Expressed in Transgenic Sugar Beet Inhibits Polygalacturonase from Rhizoctonia Solani. Biosci Biotechnol Res Asia 2011;8(1) |
| Copy the following to cite this URL: Kalantari. M, Motallebi. M, Zamani. M. R. Bean Polygalacturonase-Inhibiting Protein Expressed in Transgenic Sugar Beet Inhibits Polygalacturonase from Rhizoctonia Solani. Biosci Biotechnol Res Asia 2011;8(1). Available from: https://www.biotech-asia.org/?p=9166 |
Introduction
Most agricultural and horticultural crop species suffer from a vast array of fungal diseases which cause severe yield losses all over the world. Phytopathogenic fungi must penetrate the plant cell wall in order to initiate and expand the necrotic infections or to establish the colonization sites for biotrophic infection within the plant. Many hydrolytic enzymes produced by fungal pathogens attack the nutrient-rich polymers that constitute the plant cell wall1. Among the cell wall degrading activities produced by phytopathogens are polygalacturonases (PGs). Inhibitors of fungal enzymes that degrade the plant cell walls have been proposed to be part of the plant defenses that limit the development of disease symptoms caused by microbial pathogens2,3,4.
Polygalacturonase-inhibiting proteins (PGIPs) are the cell wall located glycoproteins that specifically inhibit the fungal PGs. They belong to a large family of the leucin-rich-repeat (LRR) – proteins4,5. By acting as both inhibitors and regulators of the PG, the PGIPs favour the release of oligogalacturonides, which are the elicitors of a variety of defense responses5. The PGIPs have been identified in the plant kingdom, and they have specific recognition abilities against many PGs produced by fungi 6,7,8.
The antifungal properties of the PGIPs were confirmed in a transgenic approach. There are several reports indicating the use of pgip genes with the target of increasing disease resistance to the fungal pathogens. Powell et al. (2000)9 and Joubert et al. (2006)10 introduced the pgip gene from pear and grapevine to tomato and tobacco respectively. They demonstrated that the inhibition of the fungal PGs slows the expansion of the disease lesions and the associated tissue maceration. Oelofse et al. (2006)11 demonstrated that the apple pgip gene expressed in the transgenic tobacco inhibits the PG of Botryosphaeia obtuse and Diaporthe ambigua which are two important pathogens of apple trees. The same results have been reported when respberry PGIP expressed in the transgenic peas was interacted with the PGs from Stenocarpella maydis and Colletotrichum lupine12.
Here we report the successful transformation of the sugar beet with the pgip2 gene from bean cultivar Emerson. The presence of the pgip gene in the transformed sugar beet was analyzed by PCR and southern dot blot. The activity of the expressed PGIP in the transgenic sugar beet was evaluated by its interaction with the polygalacturonase enzyme from Rhizoctonia solani by using agarose diffusion assay and ELISA.
Materials and methods
Plant material
To obtain the hypocotyls and leaves of Phaseolus vulgaris L. cv. Emerson, seeds (collected from Agricultural Research Centre for Seed Production, Karaj, Iran) were germinated and grown for 10 days in moist steriled soil and maintained at 25°C with a 16 h. light period.
Fungal isolates and growth conditions
One highly virulent (HV) isolate of R. solani (AG2-2) was maintained in the potato dextrose agar (PDA) at 4°C, and were grown in the shake culture on the PZ medium containing 2.64 g (NH4)2SO4, 0.34 g KH2PO4, 0.14 g MgSO4.7H2O, 10 g Citrus pectin, 1 litre dH2O. The pH adjusted to 4.513.
PGIP activity assay
To extract the PGIP containing protein three grams of the lyophilized bean hypocotyls were homogenized in 25 ml buffer containing 50 mM sodium acetate (pH 5.2), 1.5 M NaCl and stirred overnight at 4°C. After filtration through Miracloth, the insoluble tissue was re-extracted. The sodium chloride extracts were combined, centrifuged at 12000g for 30 min and the supernatant was dialyzed against 50mM sodium acetate (pH 5.2) and used as a PGIP sources14. Protein extraction from sugar beet seedling was prepared and used as negative control.
For PG extraction, the R. solani isolate was grown on 10 ml of the PZ medium in 25 ml Erlenmeyer flasks for 6 days at 21°C. The Mycelium was removed by vacuum filtration and the filtrate was clarified by centrifugation at 12000g for 5 min at 4°C. The supernatant was collected and used for the enzyme assay. Assays were repeated three times. All controls were performed using the heat-denatured enzyme.
The inhibition of the PG activity was determined by measuring the release of the reducing groups using the Somogi assay with Nelson’s arsenomolibdate reagent15 in the absence and presence of the PGIP. The PG activity was determined in 0.1 ml reaction mixture containing 0.5% (w/v) polygalacturonic acid as substrate, 50 mM sodium acetate (pH 5.2) and suitable amounts of the culture filtrates. The samples were maintained at 37°C for 60 min. One unit of the PG activity was defined as the amount of enzyme that release 1 µmol of galacturonic acid per minute. The same mixture containing the PGIP was used to assay the PGIP activity.
Cloning of the pgip2 gene
The leaf material from Phaseolus vulgaris cv. Emerson, was harvested, lyophilized and grinded into the fine powder for extraction of the genomic DNA by the method of Doyle and Doyle (1990)16. The DNA fragment containing the pgip2 gene was amplified by PCR using the genomic DNA. The primers used for amplification the pgip2 gene were designed based on the pgip2 sequence in GenBank from NCBI web site (www.ncbi.nlm.nih.gov). The pgip2 gene was amplified by PCR using the specific primers 2RB1 forward: 5′-GUGT CCT CAA GCT TAA GCA TAA TTT TG-3′ and 2RB2 reverse: 5′-GCA CGA GCT CTT AAG TGC AGG CAG GAA GAG G-3′ with the XbaI and SacI sites at the 5′ end of the primers (underlined), respectively. The PCR reactions contained 2.5 units of the Fermentas Pfu DNA polymerase, 1X buffer, 200 µM of each deoxynucleotide triphosphate, 2 µM MgSO4 and 0.5 µM primers. The reaction conditions for PCR amplification were 94°C for 90 sec, 56°C for 45 sec, and 72°C for 150 sec, for 34 cycles followed by a final extension of 5 min. The PCR products were separated by the electrophoresis on a 1% agarose gel. The PCR product (1kb) was cloned into the pUC19 plasmid and sequenced from both directions with the M13 standard primers, using the dideoxy chain termination method.
Construction of binary vector and Agrobacterium strain
The A. tumefaciens strain LBA4404 harboring the binary vector pBIAE2 harboring the pgip2 gene was used in the experiments. The plasmid pBIAE2 contains, within the T-DNA region, a neomycin phosphotranferase II (nptII) gene as the selectable marker that is a kanamycin-resistant gene for plant selection; a pgip2 gene, encoding the polygalacturonase inhibiting protein (PGIP) from Phaseolus vulgaris L. cv. Emerson. The nptII gene is regulated by the nopaline synthase promoter and terminator; the pgip2 gene is regulated by the Cauliflower mosaic virus 35S promoter (CaMV 35S) and terminated by the nos terminator.
Preparation of explants and bacterial strain for transformation
The seeds were sterilized by being submerged in 70% ethanol for 5 min and then in 0.1% HgCl2 for 8 min. They were then rinsed several times with the sterilized water and plated on the ½ MS medium17 under the light for 5 days. After germination, the cotyledonary petioles were cut and placed on the MS solid medium with 3.5mg/l benzylaminopurine (BAP)(CM-medium) for pre-culture. Two weeks later, the leaf blades which were cut from shoots were used as explants for transformation.
Single colonies of the A. tumefaciens strain harboring the pBIAE2 containing the pgip2 gene were grown in the LB medium supplemented with 50 mg/l kanamycin, and allowed to grow overnight at 27-28°C with the constant shaking (200 rpm) to mid-log phase. The bacterial culture was then transferred to a fresh medium with the amount of 0.1% and cultivated till OD600 = 0.4 with the liquid medium. The bacterial cells were collected by centrifugation and re-suspended in the ½ MS medium for use.
Transformation and selection procedure
The explants were immersed in the bacterial suspension for 1.5 min with the constant shaking, and then placed onto the sterile filter paper to remove the excessive moisture, and placed on the CM medium in the Petri dishes for co-cultivation at 25 for 2 days.
After co-cultivation, the explants were washed with the sterile water containing 200 mg/l cephatoxim to inhibit the growth of the A. tumefaciens attached to the explants and then transferred to the MS solid medium with 3.5 mg/l BAP, 15 mg/l kanamycin, and 200 mg/l cephatoxim. After shoot initiation, the explants were transferred to the MS solid medium with 25 mg/l kanamycin, and 200 mg/l cephatoxim. Regenerating shoots (about 3 cm in length) were excised from the explants and transferred to the MS solid medium with 2 mg/l 3-Indolebutyric acid (IBA), 25 mg/l kanamycin, and 200 mg/l cephatoxim for rooting and recovering the complete plants. All the above media contained 3% (w/v) sucrose with the pH 5.8, and all the explants, were cultured under 23-25°C and 16 h of day time with the high intensity of 2000 Lux.
Dot blot analysis
The genomic DNA was extracted from the fresh leaves of the putative transgenic plants and the untransformed control plant with the Cetyl Trimethyl Ammonium Bromide (CTAB) method16. The genomic DNA (5µg) was denatured for 10 min in the boiling water and chilled on ice. The denatured genomic DNAs were spotted on a nylon membrane (Hybond N+, Amersham), and hybridized to the Dig-dUTP labeled pgip probe. A fragment (1002 bp in size) was obtained from the PCR amplification of the pgip gene using 282RB1/282RB2 primers and plasmid pBIMK1 containing the pgip gene as template and subjected to the DIG DNA labeling (Roche Applied Science Gmbh, Germany) and used as a probe in the hybridization experiments.
Bioassay of transgenic sugar beet plants
The antifungal activity of the crude extract of the transgenic sugar beet was tested by using the radial diffusion assay in agarose. The canola leaf material (3 g) was ground to a fine powder in the liquid nitrogen using a mortar and pestle. Two volumes of 1 M NaCl in 20 mM NaOAc, pH 4.7 were added to the leaf material. The extracts were then shaken for 1 h at 4°C. The extracts were subsequently centrifuged at 13,000g for 20 min at 4. The pellets were discarded and the supernatants were used in the dialysis step. The samples were dialysed twice for 2 h at 4°C against 20 mM NaOAc (pH 4.7). A 12,000 MW cut-off dialysis membrane was used. The extracts were subsequently centrifuged at 13,000g for 20 min at 4°C and the supernatants stored at -20°C. The protein content was determined against BSA using the Bradford assay18. The inhibition of ploygalacturonase activity from the culture filtrate of R. solani was determined by the radial diffusion assay in agarose described by Taylor and Secor (1988)19.
Serological analyses
The PGIP level in the crude extract of transgenic sugar beet plants was determined using the ELISA. The crude proteins were incubated with 150 µl of serum (containing PGIP antibody) diluted 1:75 in PSA at 37ºC for 30 min. After washing with PBS/Tween, the antigen-antibody was incubated with 50 µl of goat anti-rabbit IgG conjugated with the horse-radish peroxidase, diluted 1000 times in PBS at 37°C for 30 min. Several washing with the PBS/Tween were then carried out. The substrate solution (100 µl), consisting 0.325% (w/v) orthophenylenediamine dihydrochloride (OPD, Sigma) and 0.085% (v/v) H2O2 was added and incubated at room temperature for 3h in the dark. The reaction was stopped with 3N HCl and the plates read in an ELISA reader at 492 nm.
Sequencing and computer analysis
The cloned DNA fragments in pUC19 (70-220 ng/µl) were sequenced by a Commercial Service (Seqlab, Gottingen, Germany). Computer analysis of the sequences was carried out and the deduced amino acid sequence from the pgip2 gene was obtained by the BLASTX Network Service (NCBI) and the multiple alignment was generated using the ClustalW (http://www.ebi-ac.uk/ClustalW).
Results and Discussion
The PGIP extract from the Phaseolus vulgaris (cv. Emerson) inhibited the PGs from the sugar beet root rot pathogen R. solani AG2-2 (69.3% PGIP inhibitory activity) when compared with the boiled PGIP which abolished the inhibition activity (Fig 1). This cultivar demonstrated the highest PGIP activity among the thirteen bean cultivars (data not shown). Boiling the extracts completely inactivated the PGIP in the extracts as indicated by comparisons of the PG inhibition by the Emerson cultivar PGIP with the inhibition of boiled PGIP. The same results have been reported by Tamura et al (2004)20, who found that boiled PGIP from persimmon demonstrated no significant inhibition of Botrytis cinerea PG. The protein extracted from the sugar beet seedlings showed no detectable inhibition of PG, suggesting that sugar beet tissue at least when developing in culture, expresses not detectable endogenous PGIP encoding gene. The finding of this study representing the significant PGIP activity indicates that the bean PGIP could be used in a strategy for the transgenic sugar beet development against the root rot disease. Meanwhile, bean plants contains a mixture of PGIPs21 and expression of the cloned pgip gene is a convenient way to study the inhibitory activity of a single pgip gene product.
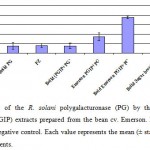 |
Figure 1: Inhibition of the R. solani polygalacturonase (PG) by the polygalacturonase-inhibiting protein (PGIP) extracts prepared from the bean cv. |
Emerson. Protein extracts from sugar beet used as negative control. Each value represents the mean (± standard error) of three independent experiments..
In this study, we isolated the pgip2 gene from the Phaseolus vulgaris cv. Emerson. The PCR amplification using the specific primers, was performed on the genomic DNA generating the specific band of approximately 1 Kb which was cloned in the pUC19 and the new construct (pUCSH1) confirmed by PCR (Fig 2), digestion patterns and sequencing. This sequenced pgip2 gene was highly similar and the coding polypeptide was identical to that of isolated independently by D’Ovidio et al. (2004)21 from P. vulgaris cv. Pinto (with accession no. AJ864507) and Hosseinzadeh et al. (2005a and b)22,23 from P. vulgaris cv. Derakhshan and cv. Naz (with accession no DQ105561 and DQ105560, respectively). The pgip2 gene encodes a protein of 333 amino acids with molecular mass of 36 kDa.
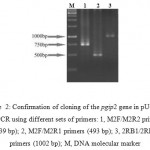 |
Figure 2: Confirmation of cloning of the pgip2 gene in pUCSH1 by PCR using different sets of primers: 1, M2F/M2R2 primers (739 bp); 2, M2F/M2R1 primers (493 bp); 3, 2RB1/2RB2 primers (1002 bp); M, DNA molecular marker. |
This sequence was inserted between the CaMV 35S promoter and the nopaline synthase terminator in the binary expression vector pBI121 and confirmed by PCR (Fig 3) and sequencing. The new construct designated as pBIMK1. Agrobacterium tumefaciens LBA4404 used for transformation of the sugar beet line 9597-p26. The success of Agrobacterium-mediated plant transformation can be a function of the genotype of the species to be transformed, the strain (virulence) of Agrobacterium, the selectable marker, the regeneration capacity of the target cells and the accessibility of the bacterium to the regenerable cells. We examined the expression of the P. vulgaris cv. Emerson pgip2 gene in the transgenic sugar beet. Also, CaMV 35S promoter used to ensure high levels of gene expression in all tissues.
 |
Figure 3a: Schematic representation of pBIMK1 and position of different primers used for PCR reaction.
|
B) Confirmation of cloning of the pgip2 gene in pBIMK1 by PCR using different sets of primers: 1, 35S/nosR primers; 2, 35S/2RB2 primers; 3, 2RB1/nosR primers; 4, 2RB1/2RB2 primers; M, DNA molecular marker
A total of 23 independent transgenic sugar beet lines were successfully rooted on the kanamycin-containing selection media (Fig 4). Approximately 57% of the regenerated plants showed to contain the pgip2 transgene (a fragment corresponding to the size “1002 bp” of the pgip2 gene) detectable PCR (Fig 5). The pgip2-specific PCR primers did not amplify a pgip in the untransformed sample.
 |
Figure 4: Transformation and regeneration of sugar beet plants. A, Shoot apices; B, Co-cultivation of inoculated explants with Agrobacterium; C, Shoot regeneration after transformation; D,
|
Regenerated shoots with well developed roots and leaves; E, Regenerated plants in pot and acclimated to non-aseptic environment; F, transgenic sugar beet plant in the green house.
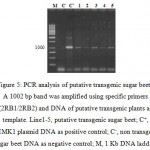 |
Figure 5: PCR analysis of putative transgenic sugar beet .
|
A 1002 bp band was amplified using specific primers (2RB1/2RB2) and DNA of putative transgenic plants as template. Line1-5, putative transgenic sugar beet; C+, pBIMK1 plasmid DNA as positive control; C–, non transgenic sugar beet DNA as negative control; M, 1 Kb DNA ladder.
Also, Southern Dot blot analyses were performed to verify the integration of the transgenes and the results reconfirmed the PCR positive plants (Fig 6). The transgenic lines were phenotypically analyzed and compared to the untransformed controls and did not show any abnormalities with regards to growth and size.
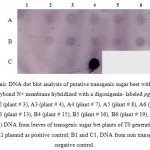 |
Figure 6: Genomic DNA dot blot analysis of putative transgenic sugar beet with the total DNA loaded onto hybond N+ membrane hybridized with a digoxigenin- labeled pgip2 probe. |
A1 (plant # 2), (A2 (plant # 3), A3 (plant # 4), A4 (plant # 7), A5 (plant # 8), A6 (plant # 10), B2 (plant # 12), B3 (plant # 13), B4 (plant # 15), B5 (plant # 16), B6 (plant # 19), C2 (plant # 21, C3 (plant # 23) DNA from leaves of transgenic sugar bet plants of T0 generation; C4, DNA from pBIMK1 plasmid as positive control; B1 and C1, DNA from non transgenic plant as negative control.
Based on the kanamycin, PCR and dot blot analysis, PGIP inhibition activity assay was performed on the selected transgenic lines by challenging the sugar beet leaf protein extracts with the PG produced in R. solani culture using the radial diffusion assay. The qualitative results demonstrated that the PGIP activity was detected in the protein extract from the leaves of the transgenic plants by inhibition of the R. solani PG (Fig 7). No PGIP activity was identified in the protein extract from the untransformed plants. The inhibition of the pathogen PGs by PGIP in vitro suggests that the plant PGIP is a deterrent to pathogen degradation of plant cell walls.
 |
Figure 7: Samples of agarose diffusion assay of crude protein extract from leaves of transgenic sugar beet plants (A and B). Wt, wild type sugar beet. |
A clear zone is indicative of polygalacturonase degrading the polygalacturonic acid. The lack of the halo indicates the inhibition of polygalacturonase activity.
Based on the PGIP inhibition activity detected by the radial diffusion assay, the expression of the recombinant PGIP in the transgenic plants (9 out of 13) was detected by ELISA using the specific PGIP antibody. Significant differences observed between the optical densities at 492 nm of the extracted proteins from the transgenic lines and the non- transformed plant as negative control (Fig 8). The variable expression of the PGIP is in agreement with the results of De Bolle et al. (2003)24 and Richter et al. (2006)12. Many factors such as the transgene localisation and the copy number25,26,27, can contribute to the variation in the transgene expression.
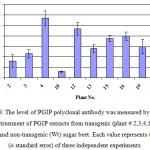 |
Figure 8: The level of PGIP polyclonal antibody was measured by ELISA after antibody treatment of PGIP extracts from transgenic (plant # 2,3,4,10,12,13,15,16, and 19) and non-transgenic (Wt) sugar beet. |
Each value represents the mean (± standard error) of three independent experiments.
There are several reports indicating the use of the pgip genes with the target of increasing disease resistance to fungal pathogens. Powell et al. (2000)9 and Joubert et al. (2006)10 introduced the pgip gene from pear and grapevine to the tomato and tobacco respectively. They demonstrated that the inhibition of the fungal PGs slows the expansion of the disease lesions and the associated tissue maceration. Oelofse et al. (2006)11 demonstrated that the apple pgip gene expressed in the transgenic tobacco inhibits the PG of Botryosphaeia obtuse and Diaporthe ambigua which are two important pathogens of apple trees. The same results have been reported when the respbarry PGIP expressed in the transgenic peas was interacted with the PGs from Stenocarpella maydis and Colletotrichum lupine12.
Transgenic techniques provide us with the probability of introducing the foreign genes into the plants to improve their resistance against fungal pathogen. In the current study, it was demonstrated that the specific product of the pgip2 gene inhibited the PGs of R. solani, an economically important pathogen, which is an important first step in disease control strategies.
Although PGIP is only one of the components of the plant resistance against pathogens and generally has only a quantitative effect on the restriction of pathogen growth, the transgenic sugar beet plants developed in this study may provide valuable material for variety improvement.
Acknowledgment
This research was financially supported by the National Institute of Genetic Engineering and Biotechnology (Grant No. 353).
References
- Elad Y., Evensen K., Phytopathology, 85: 637-643 (1995).
- Cervone F., Hahn M. G., De Lorenzo G., Darvill A. and P. Albersheim.. Plant Physiol. 90: 542-48 (1989).
- Stotz H.U., Powell A.L. T., Damon S.E., Greve L.C., Bennett A.B. and Labavitch J.M. Plant Physiol. 102(1): 133-138 (1993).
- Stotz H.U., Contos J.J.A., Powell A.L.T., Bennett A.B. and Labavitch J.M., Plant Mol Biol 25:607–617 (1994).
- De Lorenzo G., D` Ovidio R. and Cervone F., Annu Rev Phytopathol, 39:313–335 (2001).
- Yao K., De Luca V. and Brisson N., Plant Cell. 7: 1787-1799 (1995).
- Desiderio A., Aracri B., Leckie F., Mattei B., Salvi G., Tigelaar H., Van Roekel J.S.C., Baulcombe D.C., Melchers L.S., De Lorenzo G. and Cervone F., Mol Plant-Microbe Interact 10:852–860 (1997).
- Leckie F., Mattei B., Capodicasa C., Hemmings A., Nuss L., Aracri B., De Lorenzo G. and Cervone F., EMBO J 18:2352–2363 (1999).
- Powell A.L., van Kan J., ten Have A., Visser J., Greve L.C., Bennett A.B. and Labavitch J.M., Mol Plant Microbe Interact, 13:942-950 (2000).
- Joubert D.A., Slaughter A.R., Kemp G., Becker J.V.W., Krooshof G.H., Bergmann C., Benen J., Pretorius I.S. and Vivier M.A., Transgenic Res, 15:687–702 (2006).
- Oelofse D., Dubery I.A., Meyer R., Arendse M.S., Gazendam I. and Berger D.K., Phytochemistry 67:255–263 (2006).
- Richter A., De Kathen A., De Lorenzo G., Briviba K., Hain R., Ramsay G., Jacobsen H.J. and Kiesecker H., Plant Cell Rep 25: 1166–1173 (2006).
- Sweetingham M.W., Cruickshank R.H. and Wong D.H., Trans. Br. Mycol. Sci. 86: 305-311 (1986).
- Bennett A., Labavitch J. M., Powell A. and Stotz H., Regents of the university of California, Oakland, Calif., United states. (1996).
- Collmer A., Ried J. L. and Mount M. S.,. Methods in Enzymology.161: 329-335 (1988).
- Doyle J.J. and Doyle J.L., Focus 12:13–15 (1990).
- Bradford MM.. Anal. Biochem. 72, 248–254 (1976).
- Murashige T. and Skoog F., Physiol Plant 15:473–497 (1962).
- Taylor R.J. and Secor G.A., Phytopathology, 78: 1101-1103 (1988).
- Tamura M., Gao M., Tao R. and Labavitch J.M., Scientia Horticulturae, 103: 19-30 (2004).
- D’Ovidio R, Raiola A, Capodicasa C, Devoto A, Pontiggia D, Roberti S, Galletti R, Conti E, O’Sullivan D, and De Lorenzo G., Plant Physiology, 135, 2424–2435 (2004).
- Hosseinzadeh Colagar A., Zamani M.R. and Motallebi M., Direct Submission to NCBI (2005a).
- Hosseinzadeh Colagar A., Motallebi M. and Zamani M.R., Direct Submission to NCBI (2005b).
- De Bolle M.F.C., Butaye K.M.J., Coucke W.J.W., Goderis I.J.W.M., Wouters P.F.J., von Boxel N., Broekaert W.F. and Cammue B.P.A., Plant Sci 165:169–179 (2003).
- Finnegan J. and McElroy D., Biotechnology 12:883–888 (1994).
- Iyer L.M., Kumpatla S.P., Chandrasekharan M.B. and Hall T.C., Plant Mol Biol 43:323–346 (2000).
- Matzke M.A., Mette M.F. and Matzke A.J.M., Plant Mol Biol 43:401–415 (2000).

This work is licensed under a Creative Commons Attribution 4.0 International License.



