

How to Cite | Publication History | PlumX Article Matrix
A Rapid and Simple, Recombination-Based Cloning Method in Escherichia Coli
Manoj Baliram Pohare1* and Mitsuru Akita1, 2
1The United Graduate School of Agricultural Sciences, Ehime University, 3-5-7 Tarumi, Matsuyama, 790-8566 Japan.
2Graduate School of Agriculture, Ehime University, 3-5-7 Tarumi, Matsuyama, 790-8566 Japan.
Corresponding Author E-mail: b742006b@mails.cc.ehime-u.ac.jp
DOI : http://dx.doi.org/10.13005/bbra/2415

ABSTRACT: Cloning is indispensable in molecular biology. Here we developed an in vivo homologous recombination-based cloning procedure and determined the optimal conditions. This procedure required two PCR products to be amplified from a gene of interest and desired plasmid vector. The 5’ ends of both primers that amplified one product had nucleotide sequences complementary to that used to amplify the other product. Once the mixture of those PCR products was introduced into Escherichia coli DH5α competent cells, transformants carried plasmids in which the gene of interest had been properly cloned. Optimizing the cloning conditions, at least 12-nucleotides overlaps between the terminal ends of two fragments were required to generate desired plasmids. This value was much shorter than the length of overlaps required for the same procedure employed in the yeast system. Therefore, this procedure is expected to be an attractive alternative for cloning in the E. coli system.
KEYWORDS: Cloning; PCR; plasmid vector; Escherichia coli; in vivo homologous recombination
Download this article as:| Copy the following to cite this article: Pohare M. B, Akita M. A Rapid and Simple, Recombination-Based Cloning Method in Escherichia Coli. Biosci Biotech Res Asia 2017;14(1). |
| Copy the following to cite this URL: Pohare M. B, Akita M. A Rapid and Simple, Recombination-Based Cloning Method in Escherichia Coli. Biosci Biotech Res Asia 2017;14(1). Available from: https://www.biotech-asia.org/?p=21444 |
Introduction
Gene cloning is an indispensable tool in molecular biology. Conventional cloning utilizes restriction endonucleases to generate DNA fragments with complementary ends and a DNA ligase to connect these fragments prior to transformation. However, this process requires multiple steps and is time consuming. Therefore, many previous attempts have been made to overcome the limitations of the conventional cloning, e.g., the substitution of restriction endonucleases with T4 DNA polymerase or exonuclease III to generate DNA fragments,1,2 ligation-independent cloning,3,4 enzyme-free cloning,5 overlap extension PCR cloning6 and self-assembly cloning.7 However, each procedure has some disadvantages that have prevented them from replacing conventional cloning. Another cloning procedure that is free from restriction endonucleases and ligase is the Gateway Cloning Technology (Thermo Fisher Scientific Inc., Waltham, MA, USA),8 which utilizes the site-specific recombination system of a phage λ integrating in the E. coli chromosome to become a prophage. This recombination occurs at the recombination sites, attP and attB, in phage λ DNA and E. coli chromosomal DNA, respectively. In recent years, another two procedures based on in vitro homologous recombination (HR) have been developed and do not require either a restriction endonuclease digestion or a ligation step.9-12 These procedures require that the two DNA fragments to be assembled possess homologous regions at both ends. One of the procedures is called the Seamless Ligation Cloning Extract (SLiCE) method,11 which is the in vitro version of the λ prophage Red recombination system (For a review, see Ennis et al13 and involves adding λ Red α-γ overexpressed E. coli extracts to the mixture of DNA fragments. The other procedure utilizes an exonuclease of some sort. This generates the complementary single-stranded sticky ends, and subsequently, the two fragments are annealed at their ends. Then, the gap is filled in by a polymerase, and the fragments are finally ligated[9,10,12]. Enzyme kits based on this procedure have been commercialized and are available from several sources, e.g., Gibson assembly® Master Mix (New England Biolabs, Ipswich, MA, USA), In-Fusion® HD Cloning Kit (Takara Bio Inc., Kusatsu, Japan), and GeneArt® Seamless Cloning and Assembly Kits (Thermo Fisher Scientific Inc., Waltham, MA, USA). When cloning with such kits, control experiments in which the enzyme mixture was omitted often also produced transformants that carried the desired recombinant plasmids. Such an undesirable outcome served as the impetus for designing a cloning technique in which E. coli cells took up two DNA fragments and if the terminal sequence of one of the fragments was homologous to that of the other, HR could occur in vivo without any special treatment.
The cloning procedure based on in vivo HR was developed in the budding yeast Saccharomyces cerevisiae,14,15 because the organism is capable of taking up linear DNA fragments16 and recombining them efficiently[17]. This procedure is rapid and does not require the generation of cohesive ends before transformation. In fact, there are two reports in which an in vivo strategy was successfully employed in E. coli.18,19 However, in both cases, technical issues such as higher background and limited application were present. These issues have prevented this strategy from being used in a widespread manner; instead, other procedures, as described above, have been developed.
Therefore, we sought to explore whether this strategy could be more widely applicable by optimizing the experimental conditions, such as the molar ratio of two fragments and the length of overlaps between the ends of two fragments. We successfully developed a high-efficiency cloning procedure and expect that it will be an attractive alternative for cloning in the E. coli system.
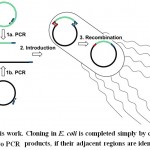 |
Sheme 1: |
Material And Methods
Enzymes, Reagents, Strain, and Oligonucleotides
The DNA polymerases for polymerase chain reaction (PCR), PrimeSTAR® Max DNA polymerase and SapphireAmp® Fast PCR Master Mix, and a restriction endonuclease Dpn I were purchased from Takara Bio Inc. (Kusatsu, Japan). The DNA ladder, HyperLadderTM 1 kb, was purchased from Bioline (London, England). DH5α E. coli competent cells were purchased from Toyobo Life Science (Osaka, Japan). The primers used in this work (Table 1) were synthesized by Eurofins Genomics Japan (Tokyo, Japan).
Table 1: List of the adjacent primers used in this study.
| Primer Name | Primer Sequence | Length of Overlap (bp) |
| pTAC2(12)_Fw
pET GFP(12)_Rv |
5’-TACAACTGATTAAGAGGGCAAAATATGAA-3’
5’-TAATCAGTTGTACAGTTCATCCATGCC-3’ |
12 |
| pET GFP(12)_Fw
pTAC2(12)_Rv |
5’-GAAGGAGATATACATATGGCTA-3’
5’-TATATCTCCTTCTTAAAGTACCG-3’ |
12 |
| pTAC2(15/21)_Fw
pET GFP(12)_Rv |
5’-CTGTACAACTGATTAAGAGGGCAAAATATGAA-3’
5’-TAATCAGTTGTACAGTTCATCCATGCC-3’ |
15 |
| pET GFP(12)_Fw
pTAC2(18)_Rv |
5’-GAAGGAGATATACATATGGCTA-3’
5’-ATGTATATCTCCTTCTTAAAGTAC-3’ |
15 |
| pTAC2(18)_Fw
pET GFP(18)_Rv |
5’-TACAACTGATTAAGAGGGCAAAATATGAAGGA-3’
5’-CCCTCTTAATCAGTTGTACAGTTCATCCATG-3’ |
18 |
| pET GFP(18)_Fw
pTAC2(18)_Rv |
5’-TAAGAAGGAGATATACATATGGC-3’
5’-ATGTATATCTCCTTCTTAAAGTAC-3’ |
18 |
| pTAC2(15/21)_Fw
pET GFP(24)_Rv |
5’-CTGTACAACTGATTAAGAGGGCAAAATATGAA-3’
5’-CCCTCTTAATCAGTTGTACAGTTCATCCATGCC-3’ |
21 |
| pET GFP(24)_Fw
pTAC2(18)_Rv |
5’-CTTTAAGAAGGAGATATACATATG-3’
5’-ATGTATATCTCCTTCTTAAAGTAC-3’ |
21 |
| pTAC2(24)_Fw
pET GFP(24)_Rv |
5’-GAACTGTACAACTGATTAAGAGGGCAAAATATGAA-3’
5’-CCCTCTTAATCAGTTGTACAGTTCATCCATGCC-3’ |
24 |
| pET GFP(24)_Fw
pTAC2(24)_Rv |
5’-CTTTAAGAAGGAGATATACATATG-3’
5’-CATATGTATATCTCCTTCTTAAAG-3’ |
24 |
| M13 forward
M13 reverse |
5’-TGTAAAACGACGGCCAGT-3’
5’-CAGGAAACAGCTATGAC-3’ |
Not Applicable |
Gene Amplification and Dpn I Treatment
Two fragments, regarded as an “insert” and a “vector,” which were used for in vivo HR, were both obtained by PCR. An “insert” fragment carrying the green fluorescent protein (GFP) gene was amplified from the plasmid, pET21b::GFP, in which the GFP gene was cloned into an expression vector, pET21b (Merck Millipore, Darmstadt, Germany). The template for the “vector” fragment was a plasmid, pTAC-2::GOI, with a synthetic gene coding for the gene of interest (GOI). Additionally, artificial 5’ untranslated region (UTR) and 3’ UTR of the GOI were cloned between the downstream of the T7 promoter and the middle of the SP6 promoter region of pTAC-2 (BioDynamics Laboratory Inc., Tokyo, Japan) via the in vivo HR method described in this paper (Figure 1). The partial DNA sequence of this plasmid is shown in Figures 1‒3.
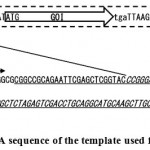 |
Figure 1: Partial DNA sequence of the template used for a vector fragment.
|
The synthetic DNA (indicated by the rectangle with a broken line) consists of the gene of interest (GOI), with both 5’and 3’ UTR inserted between the multi-cloning site, and the middle of the SP6 promoter (PSP6) of a TA vector, pTAC-2, generated by the in vivo HR method described in this article. The sequence of the multi-cloning site of pTAC-2 is underlined. The sequence of pTAC-2 that was substituted by the synthetic gene is italicized. The T7 promoter is shown as “PT7.” The nucleotide sequence of the respective areas is shown in Figure 3, line 1.
Amplification of the insert and vector fragments was carried out with PrimeSTAR® Max DNA polymerase, a set of 0.3 μM primers (Table 1) and a template (1 ng) in a GS-482 thermal cycler (G-Storm, Somerset, UK). To amplify the vector and insert fragments with 18-nucleotide (nt) overlaps, the following template DNA and primer sets were added: for the vector, pTAC-2::GOI was used as the template and pTAC2(18)_Fw and pTAC2(18)_Rv were used as primers, whereas for the insert, pET21b::GFP was used as a template and pET GFP(18)_Fw and pET GFP(18)_Rv were used as primers (Figure 2 and Table 1). All reactions were performed in 20 μl reaction mixtures under the following conditions: 40 cycles of denaturation (98°C for 10 s), annealing (55°C for 15 s), and extension (72°C for 5 s (insert) or 15 s (vector)), with the lid heated at 110°C. Five units of Dpn I, which was used to digest the methylated template, were added to the PCR amplified products (20 μl), without changing the buffer to the optimal buffer for Dpn I, and incubated at 37°C for 1 h. The Dpn I-treated PCR products were purified using the FastGene Gel/PCR Extraction Kit (Nippon Genetics Co. Ltd., Tokyo, Japan) and eluted from the column in 50 μl of H2O that was preheated to 65°C. The concentration of the purified PCR products was estimated by comparing their intensity to that of the DNA ladder, HyperLadderTM 1 kb, after the samples were separated by agarose gel electrophoresis, and the gels were stained with ethidium bromide.
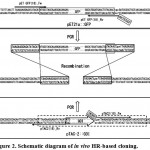 |
Figure 2: Schematic diagram of in vivo HR-based cloning.
|
We developed an in vivo HR-based cloning procedure. This procedure required two PCR products to be amplified from a gene of interest (“Insert Fragment”) and desired plasmid vector (“Vector Fragment”) only if the 5’ends of both primers that amplified one product had nucleotide sequences complementary to that used to amplify the other product. Once these fragments were co-introduced into E. coli cells, HR occurred between both fragments at the overlapping ends to complete plasmid construction. Overlaps of 18 nt are shown here as an example. Using the primers pET GFP(18)_Fw and pET GFP(18)_Rv, an insert fragment was amplified from pET21::GFP. Using the primers pTAC2(18)_Fw and pTAC2(18)_Rv, a vector fragment was amplified from pTAC-2::GOI. The nucleotide sequence of each primer is identical to the sequence below or above the single- or double-lined arrow. In addition, in order to carry the identical sequence at each end of both fragments, the 5’ ends of the primers, pET GFP(18)_Rv (5’-CCCTCTTAA-3’) and pTAC2(18)_Fw (5’-TACAAC-3’), were attached to the nucleotide sequences taken from pTAC-2::GOI and pET21a::GFP, respectively, which were in frame. The single underline indicates the nucleotide sequence taken from pTAC-2::GOI, while the double underline indicates the nucleotide sequence taken from pET21a::GFP. Overlapping sequences from both the insert and the vector fragments are italicized. The directions of the primers are indicated by the arrowheads. Overlapping ends of each fragment are italicized and indicated by the rectangles.
Transformation and Colony Counting
A PCR amplified vector fragment of 2.7 kbp (4.15 fmol) and a PCR amplified insert fragment of 0.75 kbp were combined at a molar ratio of vector:insert of 1:1, 1:2, or 1:5 and dried in the vacuum dryer, Micro VacTM MV-100 (TOMY Digital Biology Co. Ltd., Tokyo, Japan). After resuspending the mixture in 1 μl of water, a 10 μl aliquot of DH5α competent cells was added, and the samples were kept on ice for at least 20 min. Following a heat shock of 45 s at 42 °C, the samples were kept on ice for 2 min, then incubated at 37 °C for 1 h after addition of 50 μl of SOC medium. A half aliquot of the bacterial suspension was spread on LB agar plates containing 50 μg/ml carbenicillin, and the plates were incubated overnight at 37 °C. The number of colonies that appeared on plates was counted under illumination with UV light at 365 nm.
Colony PCR and DNA Sequencing
Recombination was first verified by colony PCR, with a pair of the “M13 forward” primer and the “M13 reverse” primer (Table 1), both of which annealed to the upstream of the T7 promoter and the downstream of the SP6 promoter in pTAC-2, respectively. Colonies were transferred with a sterile toothpick from a master plate into PCR tubes containing 30 μl of sterile distilled water, and E. coli cells were lysed in the thermal cycler for 5 min at 94 °C. One microliter from this E. coli lysate was used as a template for the 20 μl PCR reaction with SapphireAmp® Fast PCR Master Mix, and PCR was performed according to the manufacturer’s instructions. The PCR products were subsequently analyzed on an agarose gel.
After isolating plasmids from transformants using the FastGene Plasmid Mini Kit (Nippon Genetics Co., Ltd., Tokyo, Japan), the plasmid DNA was sequenced by Fasmac Co. Ltd. (Atsugi, Japan). Sequence alignment was performed using the Genetyx ver. 10 software (Genetyx Co., Tokyo, Japan).
Results
PCR Amplification and Cloning
To simplify the cloning process, we prepared two PCR-amplified DNA fragments that were designated as the “vector” and the “insert” by using primers with 5’ overhangs, thereby producing DNA fragments identical at their respective terminal ends (Table 1 and Figure 2). Once the sizes and purity of both PCR products were confirmed by agarose gel electrophoresis, they were treated with a restriction endonuclease Dpn I to digest the templates used for amplification. After they were purified using a kit, each fragment was quantified. Because our initial control experiments were done with fragments containing around 18-nt overlaps and using a 1:2 molar ratio of the vector to insert, we first attempted to identify the optimal ratio of the two fragments required for efficiently obtaining correct transformants by mixing 4.15 fmol of the vector with the insert at ratios of 1:1, 1:2, and 1:5.
On the agar plates, two types of colonies, green and white, were observed. The differences in these colonies were more evident when the plate was illuminated with UV light (data not shown). Because E. coli DH5α does not carry the T7 RNA polymerase gene, it is highly possible that GFP was transcribed by the endogenous E. coli RNA polymerase. Promoter prediction software, such as BPROM[20] and PromoterHunter[21] identified the nucleotide sequence overlapping with the T7 promoter recognized by sigma 70 (Figure 3A, line 1). When the respective region (indicated by the rectangle with a solid line in Figure 3A, line 1) was deleted, no green colonies appeared (data not shown). In addition, when plasmids isolated from these E. coli cells were introduced into E. coli BL21(DE3) cells, GFP was expressed in the presence of isopropyl-β-D-thiogalactoside (data not shown). From these observations, the deleted area contained elements critical for the endogenous E. coli RNA polymerase-dependent transcription as predicted. Therefore, we assumed that green colonies contained the desired plasmid.
The number of total colonies increased if the amount of the insert was doubled, and remained the same when the molar ratio was 1:5 (Table 2). However, the number of white colonies increased when the molar ratio was 1:5 as compared with 1:2, suggesting that the excess amount of insert did not improve the efficiency of obtaining the desired plasmid. Therefore, we decided to mix 4.15 fmol of the vector and 8.3 fmol of the insert in our subsequent experiments.
Table 2: Relationship between the molar ratio of the two fragments and the number of transformants.
|
Molar ratio (Vector:Insert) |
Number of colonies
[cfu (colonies / μg)] |
||
| Total | Green | White | |
| 1:1 | 9.7 ± 12.4 [(1.31 ± 1.68) × 103] | 7.3 ± 10.1 [(0.99 ± 1.37) × 103] | 2.3 ± 2.3 [(0.32 ± 0.31) × 103] |
| 1:2 | 14.0 ± 6.2 [(1.89 ± 0.84) × 103] | 10.7 ± 6.8 [(1.44 ± 0.92) × 103] | 3.3 ± 0.6 [(0.45 ± 0.08) × 103] |
| 1:5 | 13.0 ± 10.6 [(1.76 ± 1.43) × 103] | 7.0 ± 4.4 [(0.95 ± 0.59) × 103] | 6.0 ± 6.2 [(0.81 ± 0.84) × 103] |
Notes: The number of green colonies and white colonies that grew on the agar plate were counted. From the number of colonies, the colony forming unit (cfu) value (colonies /μg vector) was calculated. The average and standard deviation from three independent experiments are provided in this table.
Variation in the overlap Length
Next, we sought to address whether the length of overlaps between the two terminal ends of the vector and the insert would affect the efficiency of in vivo HR. PCR primers with different overlap lengths, from 12 to 24 nt, were prepared (Table 1). As described above, 4.15 fmol of the vector fragment and 8.3 fmol of the insert fragment were mixed and introduced into E. coli DH5α cells. We enumerated the number of green colonies obtained with the varying lengths of overlaps (Table 3). The number of green colonies increased as the length of overlaps increased. However, the efficiency of obtaining green colonies reached a plateau when the overlap length was 18 nt. Although green colonies appeared when 12-nt overlaps were used, this condition yielded the minimum number of colonies, and the ratio of green to white colonies was very low (8.3 %). From these results, we concluded that for this procedure, overlaps of more than 15 nt at both ends were necessary to obtain green colonies at a certain efficiency.
Table 3. Relationship between the length of the overlaps between two fragments and the number of transformants.
|
Overlap length (nucleotides) |
Number of colonies
[cfu (colonies / μg)] |
Green colonies /
Total colonies (%) |
||
| Total | Green | White | ||
| 12 | 24.0 ± 12.0 [(3.24 ± 1.62) × 103] | 2.0 ± 3.5 [(0.47 ± 0.27) × 103] | 22.0 ± 9.2 [(2.97 ± 1.24) × 103] | 8.3 |
| 15 | 21.3 ± 8.3 [(2.88 ± 1.13) × 103] | 9.3 ± 3.1 [(1.26 ± 0.41) × 103] | 12.0 ± 5.3 [(1.62 ± 0.72) × 103] | 43.8 |
| 18 | 28.0 ± 16.4 [(3.78 ± 2.21) × 103] | 20.0 ± 12.2 [(2.70 ± 1.64) × 103] | 8.0 ± 5.3 [(1.08 ± 0.72) × 103] | 71.4 |
| 21 | 40.7 ± 16.2 [(5.45 ± 2.18) × 103] | 28.7 ± 16.2 [(3.87 ± 2.18) × 103] | 12.0 ± 10.6 [(1.62 ± 1.43) × 103] | 70.5 |
| 24 | 51.3 ± 23.2 [(6.94 ± 3.13) × 103] | 39.3 ± 22.5 [(5.32 ± 3.04) × 103] | 12.0 ± 2.0 [(1.62 ± 0.27) × 103] | 71.6 |
Notes: The number of green colonies and white colonies that grew on the agar plate were counted. The average and standard deviation from three independent experiments were calculated. From the number of colonies, the cfu value (colonies /μg vector) was calculated. The ratio of the number of green colonies versus the number of total colonies is also shown.
Verification of Transformants by Sequencing
We classified transformants based on the color of the colonies, which indicated GFP expression in the transformants. However, GFP expression did not prove whether correct recombination between the fragments took place. Therefore, we verified the transformants by colony PCR and DNA sequencing. First, colony PCR was performed on selected green colonies with a set of M13 forward and M13 reverse primers, which annealed upstream of the T7 and SP6 promoters, respectively (Figure 3). The PCR product migrated at the expected size (data not shown). Plasmids were isolated from green colonies and sequenced. The sequencing analysis showed that in vivo HR took place at the overlapping sequences, as intended (Figure 3).
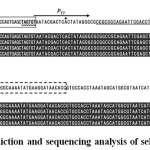 |
Figure 3: Promoter prediction and sequencing analysis of selected plasmids.
|
The sequences of the 5’ UTR (A) and 3’ UTR (B) of the template plasmid for a vector fragment (line 1) are shown. The transcription start site (indicated by a dot in the middle of the T7 promoter (PT7)) and transcription elements, such as ‒35 (double underline) and ‒10 (wavy underline) hexamers recognized by sigma 70 in the 5’ UTR, identified by promoter prediction software are also shown (Panel A, line 1). The region for deleting these transcription elements is indicated by the rectangle with a solid line. Selected plasmids, which were found to contain the insert by colony PCR, were isolated from green (lines 2‒5) colonies, and DNA was sequenced with the internal GFP primers (lines 2‒5). The sequences of the 5’ UTR (A) and 3’ UTR (B) of the GFP gene are aligned and shown as outlined. The meanings of the rectangles are as described in Figure 1.
Discussion
The in vivo HR cloning strategy has been applied in the budding yeast system for quite some time.14 However, this strategy had not been further developed in the E. coli system since 1993.18,19 As aforementioned, higher background and limited application of this strategy have prevented its widespread adoption. In the article by Bubeck et al,18 a restriction endonuclease digested vector (2.9 kbp) and a PCR amplified insert fragment (1.1 kbp), with respectively 42-nt and from 0 to 33-nt overlaps at each end were co-introduced into E. coli cells at the ratio of 1:2.6. Yields of positive transformants increased as the length of the overlaps increased. However, the rate of positive transformants did not even reach 40 %. In addition, the highest number of negative colonies appeared even with a 0-nt overlap at one end. Since the preparation of the insert fragment was not documented,18 this outcome led us to speculate the insufficient removal of a PCR template used for the insert fragment, which encoded the same antibiotic resistant gene carried by the vector fragment. To avoid such a problem, Oliner et al19 applied a restriction endonuclease digested vector to be gel purified and an insert fragment to be amplified from a template which was either a plasmid carrying a different antibiotic marker from a vector fragment or genomic DNA. However, this modification does not allow every type of DNA to be utilized as a template. Therefore, the applicability of this strategy is also limited. As such, we added Dpn I to degrade the remaining DNA templates and prevent them from being introduced into E. coli. Dpn I is known to digest methylated DNA at its recognition site.22 Therefore, all of the plasmids used for the PCR templates in this study were isolated from a dam+ E. coli strain, such as DH5α.
To adapt our procedure for broader application, we optimized two aspects of the reaction conditions: the ratio of the two fragments and the length of overlaps between the ends of the two fragments. The total number of colonies maximized at the 1:2 and 1:5 vector to insert molar ratios. However, the ratio of 1:2 was found to yield transformants most effectively (Table 2). The decrease in yield at 1:5 can be explained as follows. When the insert fragment is in excess, the possibility of two insert fragments recombining with each end of the same vector fragment may increase, which limited the availability of the intact vector fragment. As a result, the total number of positives will decrease.
It was also found that for successful cloning, the minimum number of complementary overlaps was 12-nt (Table 3). The number of positive colonies obtained increased with longer lengths of overlaps (Table 3). However, the efficiency did not further increase with overlap lengths of more than 18 nt. Because longer overlaps directly affect the cost of primer synthesis, to minimize cost, we usually design primers with overlaps of 15 to 20 nt. In addition to optimization, we applied the PCR enzyme that does not add an extra adenine nucleotide at the 3’ end to avoid possible mutation introduced at the recombination site.
To mechanistically explain our cloning procedure, homologous recombination is at the core of this process. The E. coli strain used in our experiments, DH5α, was recA negative,23 suggesting the involvement of a pathway other than RecA-dependent recombination. RecA-independent recombination was observed when 25-nt overlap was attached[24]. This overlap in length, when compared with the much longer overlap required for RecA-dependent recombination, is still longer than the minimum number of overlap (12-nt) length required for in vivo HR in our procedure. Furthermore, in this pathway, recombination takes place inside closed circular structures like the bacterial chromosome or plasmid[25,26]. In contrast, in our case, both DNA fragments were linear with the terminal sequence of one of the fragments homologous to that of the other. Therefore, RecA-independent recombination is less likely to take place. Since double-stranded DNA specific exonucleases, such as RecE[27], are present in E. coli cells, once a vector and an insert fragment are introduced in cells after heat shock treatment, single-stranded DNA overhangs will be created at both ends of the fragments. Single stranded overhangs favor annealing of these fragments in vivo and the gap is filled in by a polymerase, followed by being sealed by a ligase. This possibility may be supported by previous reports where RecE improved the recombination efficiency in E. coli.28-30 Thus, we propose a mechanism which operates in vivo and functions similarly to that of the SLiCE method,11 as the RecE recombination system is analogous to the λ Red system.31 Although we did not further evaluate other factors which might affect cloning, such as the size of fragments and GC content of overlapping sequence, we successfully generated several dozen plasmids by the in vivo HR cloning method reported herein.32,33 So far, we successfully obtained vectors (up to 8 kbp) and inserts (up to 3 kbp) via PCR from E. coli chromosomal DNA, budding yeast total DNA, Arabidopsis cDNA, and plasmids as templates (data not shown). GC content of overlapping sequence from our study varied from 25 % (pTAC(12)_Fw and pET GFP(12)_Rv) to 50 % (pTAC(15/21)_Fw and pET GFP(24)_Rv) (Table 1). On the other hand, among those plasmids we have already generated, GC content was between 5.9 % and 94.1 % (data not shown). Therefore, GC content of overlapping sequence may not be critical. However, we occasionally encountered poor efficiency. As the DNA quantitation methods were crude, the fragment mixture may not have been prepared properly. Therefore, we mixed the fragments at various vector to insert fragment ratios, which typically improved the efficiency of the technique (data not shown).
conclusion
Here, we report a time-saving, cost-effective, and simple cloning technique that was serendipitously discovered. We found that desired transformants may be obtained when a mixture of two DNA fragments containing a certain length of identical nucleotide sequences at their terminal ends is introduced into E. coli cells. Because this technique is based on homologous recombination in vivo, cloning is independent of restriction endonuclease recognition sites at the cloning site, and a gene of interest can thus be cloned into plasmid at the desired site without additional nucleotide sequences. Therefore, most of the time-consuming steps required in classical cloning protocols are avoided. In addition, because recombination occurs in vivo, enzymatic cloning materials are not required, with the exception of a DNA polymerase for PCR and Dpn I.
Acknowledgments
We thank Dr. K. Akiyama (Ehime University) for generously providing the plasmid, pET21b::GFP. This work was supported by The United Graduate School of Agricultural Sciences, Ehime University, under the Student Research Project Creation Support Program, 2014-15. This work was carried out at the Division of Genetic Research, the Advanced Research Support Center, Ehime University. M. B. Pohare is a recipient of a scholarship from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Kaluz S., Kölble K., Reid K. B. M. Directional cloning of PCR products using exonuclease III. Nucleic Acids Res. 1992;20(16):4369.
CrossRef - Stoker A. W. Cloning of PCR products after defined cohesive termini are created with T4 DNA polymerase. Nucleic Acids Res. 1990;18(14):4290.
CrossRef - Aslanidis C., de Jong P. J. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 1990; 18(20):6069–
CrossRef - Yang Y. S., Watson W. J., Tucker P. W.,Capra D. Construction of recombinant DNA by exonuclease recession, Nucleic Acids Res. 1993;21(8):1889–93.
CrossRef - Tilett D., Neilan B. A. Enzyme-free cloning: a rapid method to clone PCR products independent of vector restriction enzyme sites. Nucleic Acids Res. 1999;27(19):e26.
- Bryksin A.V., Matsumura I. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. BioTechniques. 2010;48(6):463–5.
CrossRef - Matsumoto A., Ito T. Q. Self-assembly cloning a rapid construction method for recombinant molecules from multiple fragments. Biotechniques. 2011;51(1):55.
CrossRef - Hartley J. L., Temple G. F., Brasch M. A. DNA cloning using in vitro site specific recombination. Genome Res. 2000;10(11):1788–95.
CrossRef - Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A., Smith H. O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Methods. 2009;6:343–5.
CrossRef - Hamilton M. D., Nuara A. A., Gammon D. B., Buller R. M.,Evans D. H. Duplex strand joining reactions catalyzed by vaccinia virus DNA polymerase. Nucleic Acids Res. 2007;35(1):143.
CrossRef - Zhang Y., Werling U. and Edelmann W., SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res., 2012; 40(8): e55.
CrossRef - Zhu, B., Cai, G., Hall, E.O., Freeman, G.J. In-fusion assembly: seamless engineering of multidomain fusion proteins, modular vectors, and mutations. Biotechniques, 2007; 43(3): 354–
CrossRef - Ennis, D.G., Amundsen, S.K., Smith, G.R. Genetic functions promoting homologous recombination in Escherichia coli: a study of inversions in phage lambda. Genetics, 1987; 115(1): 11–
- Ma, H., Kunes, S., Schatz, P.J., Botstein, D. Plasmid construction by homologous recombination in yeast. Gene, 1987; 58(2-3): 201–16.
CrossRef - Oldenburg, K.R., Vo, K.T., Michaelis, S., Paddon, C. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res., 1997; 25(2): 451–2.
CrossRef - Hinnen, A., Hicks, J.B., Fink, G.R. Transformation of yeast. Natl. Acad. Sci. USA, 1978; 75(4): 1929–33.
- Orr-Weaver, T.L., Szostak, J.W., Rothstein, R.J. Yeast transformation: A model system for the study of recombination. Natl. Acad. Sci. USA, 1981; 78(10): 6354–8.
CrossRef - Bubeck, P., Winkler, M., Bautsch, W. Rapid cloning by homologous recombination in vivo. Nucleic Acids Res., 1993; 21(15): 3601–2.
CrossRef - Oliner, J.D., Kinzler, K.W., Vogelstein, B. In vivo cloning of PCR products in coli. Nucleic Acids Res., 1993; 21(22): 5192–7.
CrossRef - Solovyev, V., Salamov, A. Automatic Annotation of Microbial Genomes and Metagenomic Sequences. In: Li RW, editor. Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies, Hauppauge (NY): Nova Science Publishers, 2011; 61–Klucar, L., Stano, M., Hajduk, M. phiSITE: database of gene regulation in bacteriophages. Nucleic Acids Res., 2010; 38: D366–D70.
CrossRef - Russell, D.W., Zinder, N.D. Hemimethylation prevents DNA replication in coli. Cell, 1987; 50(7): 1071–9.
CrossRef - Casali, N. Escherichia coli host strains. Methods Mol. Biol., 2003; 235: 27–48.
CrossRef - Lovett, S.T., Hurley, R.L., Sutera, V.A.Jr., Aubuchon, R.H., Lebedeva, M.A. Crossing over between regions of limited homology in coli. RecA-dependent and RecA-independent pathways Genetics, 2002; 160(3): 851–9.
CrossRef - Dutra, B.E., Sutera, V.A.Jr., Lovett, S.T. RecA-independent recombination is efficient but limited by exonucleases. Natl. Acad. Sci. USA, 2007; 104(1): 216–21.
CrossRef - Swingle, B., Markel, E., Costantino, N., Bubunenko, M.G., Cartinhour, S., Court, D.L. Oligonucleotide recombination in Gram-negative bacteria. Microbiol., 2010; 75(1): 138–48.
CrossRef - Joseph, J.W., Kolodner, R. Exonuclease VIII of coli. I. Purification and physical properties. J. Biol. Chem., 1983; 258(17): 10411–7.
- Keim, P., Lark, K.G. The RecE recombination pathway mediates recombination between partially homologous DNA sequences: Structural analysis of recombination products. Struct. Biol., 1990; 104(1-3): 97–106.
CrossRef - Symington, L.S., Morrison, P., Kolodner, R. Intramolecular recombination of linear DNA catalyzed by the Escherichia coli RecErecombination system. Mol. Biol., 1985; 186(3): 515–25.
CrossRef - Yamaguchi, H., Hanada, K., Asami, Y., Kato, J.I.,Ikeda, H. Control of genetic stability in coli: the SbcB 3’-5’ exonuclease suppresses illegitimate recombination promoted by the RecE 5’-3’ exonuclease. Genes to Cells, 2000; 5(2): 101–9.
CrossRef - Gillen, R.,Karu, A.E., Nagaishi, H., Clark, A.J. Characterization of the deoxyribonuclease determined by lambda reverse as exonuclease VIII of E. coli. J. Mol. Biol., 1977; 113: 27–41.
CrossRef - Pohare, M.B., Akita, M. Development of a precursor in a soluble form for protein import into chloroplasts. IJBTR, 2016; 6(5): 9–
- Akita, M., Pohare, M.B. Chloroplastic protein import characteristics of dihydrofolate reductase (DHFR) fused recombinant precursor protein in the presence of methotrexate. , Biotechnol. Res. Asia, 2016; 13(4): (In press).
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.



