

How to Cite | Publication History | PlumX Article Matrix
Analysis of Gene Expression using Modified Relative Codon Bias Strength in Nanoarchaeum Equitans
Shibsankar Das
Department of Mathematics, Uluberia College, Uluberia, Howrah, West Bengal, India-711315.
DOI : http://dx.doi.org/10.13005/bbra/2510

ABSTRACT: Genes expressed at higher level exhibit extreme codon bias towards the use of optimal set of codons and based on this hypothesis, several codon usage indices are used to predict gene expressions in different genomes. This study analyzed a codon usage methodology, MRCBS (Modified Relative Codon Bias Strength) to explicate the outlines of gene expression in the Nanoarchaeum equitans kin4-M genome. The analysis of this work revealed that most of the highly expressed genes of this genome involved in protein synthesis, DNA/protein metabolism, protein fate, cellular process, ribosomal / translation and transcription factor, amino acid biosynthesis and many hypothetical proteins. It is also observed that MRCBS correlates better with codon adaptation index than the other widely accepted codon bias measures.
KEYWORDS: Codon bias index; codon bias; CAI (Codon Adaptation Index)GC content; predicted highly expressed genes;
Download this article as:| Copy the following to cite this article: Das S. Analysis of Gene Expression using Modified Relative Codon Bias Strength in Nanoarchaeum Equitans. Biosci Biotech Res Asia 2017;14(2). |
| Copy the following to cite this URL: Das S. Analysis of Gene Expression using Modified Relative Codon Bias Strength in Nanoarchaeum Equitans. Biosci Biotech Res Asia 2017;14(2). Available from: https://www.biotech-asia.org/?p=24673 |
Introduction
Predicting expression level of a gene from nucleotide sequence remains an exciting problem in the field of modern bioinformatics. Gene expression in micro-organism is controlled by a number of factors which are participated in protein biosynthesis, mutation, and folding. The important feature of gene expression is observed that codon usage bias varies strongly between genes which are expressed at higher level than genes expressed at lower level and this biased amino acid composition designate the biological importance of these protein coding genes. Recently there have been considerable studies to calculate the genes expressed at higher level in prokaryotes and in eukaryotes and a range of software tools emarged to study gene expression. Comprehensive experimental approaches like cDNA microarray, SAGE etc. require a massive investment of time and resources and put an obstacle before the investigators. Thus, to predict the genes expressed at higher level in most of the genomes, scientists were started to think use of computational methods instead of direct experimental methods. So using computational methods instead of experimental approaches in calculating expression profile of the genome becomes exciting because it avoids luxurious and time-consuming experimentation.
It is postulated that highly expressed genes preferably choose an optimal subset of codons and often are depicted by strong codon usage bias,1-8 several gene expression measures like codon adaptation index (CAI), 9 Relative Codon Adaptation (RCA),10 Relative Codon Bias Strength (RCBS)11-13 are available to compute codon bias of the genes in the literature. Both CAI and RCA use the codon usage of highly expressed genes as reference set and so determination of reference set of highly expressed gene is main disadvantage in calculation of CAI and RCA. Even though the identification of reference set is not necessary for computing score of RCBS, but RCBS has partial dependency on length of genes (for genes having length < 300 aa).10 In order to get rid of the problems of partial dependence on gene length and determination of reference set, another model (MRCBS) is proposed. MRCBS uses Ribosomal Protein (RP) genes which are supposed to be genes expressed at higher level3,4 as a reference set, to predict the expression levels of genes and has no length dependency.
Nanoarchaeum equitans kin4-M was revealed in 2002 by Karl Stetter in a hydrothermal vent off the coast of Iceland on the Kolbeinsey Ridge. It is a species of marine thermophilic Achaea that nurtures at about 800C in temperature. Thermophilic organisms exhibit more bioleaching rate than non-thermophilic organisms at high temperature, so thermophilic microbes create interest in the application of biomining industry. Genes essential for synthesis of amino acids, nucleotides, cofactors, and lipids are almost absent in small genome of Nanoarchaeum equitans (490,885 base pairs), but translates genes required for repair and replication. This limitation of synthesis of amino acids shows that N. equitans is related to archaeal parasite, Ignicoccus. Nanoarchaeum equitans can nurture only with host archaeon, Ignicoccus sp., and phylogenetic analysis of their 16S rRNA sequences suggests that they separated early in the archaeal lineage, before the development of the Euryarchaeota and Crenarchaeota. This symbiosis and remarkable evolutionary divergence of N. equitans raised the interest among evolutionary biologist on whether it has gone through extensive evolution.14,15 Thus, the gene expression of N. equitans is expected to make important contributions in several areas. In this work, the gene expression and the variation in patterns of synonymous codon usage across the genes of Nanoarchaeum equitans kin4-M using MRCBS is investigated. To measure the performance of the proposed codon usage measure, MRCBS is compared with the performances of several commonly used measures.
Materials and Methods
The whole genome of Nanoarchaeum equitans kin4-M along with gene annotations is retrieved from NCBI GenBank. In this study, all coding DNA sequence (CDS) which are annotated as hypothetical and predicted by computational methods has been used. Nanoarchaeum equitans kin4-M (400 nm in diameter and 31.56% GC content), smallest known Achaean encodes 536 proteins, 82% of which are annotated as either “hypothetical protein” or “protein of unknown function”). In the following sections, the different expression level predictors CAI, RCBS, RCA, GC3 and Impact Codon is briefly described and an alternative approach using the score of modified relative codon bias strength (MRCBS) is explained.
Codon Adaptation Index (CAI) [9]
The codon adaptation index (CAI) calculates the difference of codon usage of a particular gene from the codon usage of the highly expressed genes in the reference set. Let fi,aa is the frequency of codon i, and faa,max is the maximum frequency of the codon used for encoding amino acid aa in a set of highly expressed genes. The relative adaptiveness (wi) is obtained by normalizing the frequency of each synonymous codons by the frequency of the most frequently used codon i.e.
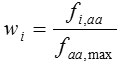
The CAI of a gene is computed as the the geometric mean of wi ‘s of all codons (excluding stop codons) in a gene sequence of length N,
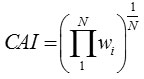
The values of CAI range from 0 to 1 and genes having values close to 1 are supposed to be PHE genes.
Relative Codon Adaptation (RCA) [10]
Fox et al.10 suggested RCA which computes codon bias of a gene using codon bias of the highly expressed genes in the reference set. Relative adaptation for individual codon xyz is defined as
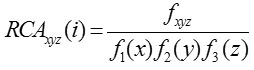
Using this, RCA for entire gene is computed as
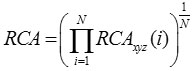
where fxyz is the relative frequency of codon xyz in any reference set of highly expressed genes, Fm (n)is the relative occurrence of nucleotide base n at codon position m in any reference set of highly expressed genes and N is the number of codons in the particular gene sequence.
Relative Codon Bias Strength (RCBS)[11-13]
The codon usage index of a gene designated by RCBS is calculated as
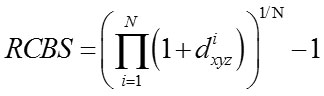
and
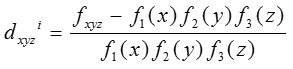
where fxyz is the relative frequency of codon xyz and Fm (n)is the relative occurrence of nucleotide base n at codon position m and N is the number of codons in the particular gene sequence.
GC3
Highly expressed genes always use a subset of favored codons. These biased codons are either C or T ending at the third position. C. Shields et al.16 established that GC contents were frequently interrelated with expression patterns of genes. The GC3 content is calculated as the frequency of G or C nucleotides at the third position of codons except Met, Trp and stop codons i.e.
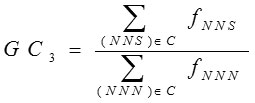
Where N =any base, S=G or C. and fxyz is the observed frequency of codon xyz
Impact Codon
Das et al.17 calculated the gradation of codon bias of every codon by computing ana score, denoted as impact score (dxyzi as it reflects codon usage bias as well as the compositional bias of nucleotide base. Impact score is defined as
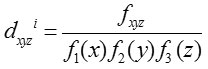
Where fxyz is the relative frequency of codon xyz and Fm (n) is the relative occurrence of nucleotide base n at codon position m in a particular gene. Let
![]()
and µ represent the sample mean and population mean of dxyzi respectively; and σ the population standard deviation, then z score is calculated as
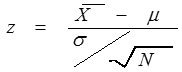
where N is the gene length. Based on the z score of each codon, a codon is classified as impact codon if its z score qualifies significance level (general used 5% significance level).
Modified Relative Codon Bias Strength (MRCBS) [18-19]
The proposed method is interrelated to codon usage of a gene with respect to compositional bias of nucleotide base at the three codon sites. Let fxyz be the calculated normalized codon frequency for the codon triplet (x,y,z) in a particular reference set of gene and Fm (n) is the relative observed frequency of base m at codon position n in the same reference set.
Thus the codon usage measure of a gene is defined as
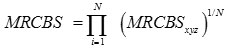
where
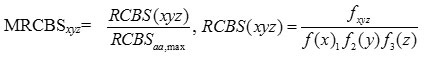
RCBSaa, max is the maximum RCBS of codon encoding same amino acid aa in the same reference set, and N is the length in gene. MRCBxyz is independent of the size of the reference set as it is the ratio of the RCBS of the codon xyz to the maximum of RCBS of codon encoding same amino acid. The value of MRCBS lies between 0 and 1.
In this study, the criteria MRCBS > T (Threshold score) was taken as a standard for recognizing the highly expressed genes and methodology used to calculate threshold score as described in Sahoo and Das.19 Due to variation of codon usage bias as well as compositional nucleotide bias, threshold score for identifying highly expressed genes varies from genome to genome. For calculating threshold score (T), a parameterization set to evaluate how well CAI value correlates actual expression level predicted by MRCBS is defined. First, CAI is calculated for all the genes by taking all RP (>80 aa) genes as highly expressed genes and called this as an evaluation set. Then taking a random threshold score T of MRCBS, highly expressed genes is differentiated from expression of gene at lower level. The genes expressed at higher level with expression level (MRCBS) greater than T define parameterization set. Relative adaptiveness of CAI is computed by taking parameterization set as reference set. With the parameters in hand, CAI scores of all the sequences in the data set were computed. In order to evaluate how well the MRCBS predicts expression level, the correlation between CAI values of the data set and evaluation set is calculated and denoted as merit of the method. The process is iterated by varying the threshold T and repeating the procedure till optimal merit of the method is reached. This way, the optimal threshold value of MRCBS of the whole genome expression data is obtained.
In this work, codon usage measures CAI, RCA and MRCBS have been calculated by relating its codon usage bias with the codon usage of the housekeeping genes, for example the ribosomal protein (RP) genes, which are commonly believed to PHE genes. The PHE are then categorized based on the degree of the codon usage bias obtained from measures as defined above and a gene is recognized as highly expressed gene if its MRCBS exceeds threshold value T. All the correlation used in this work is the Pearson r correlation coefficients between different codon usage bias indices computed.
Results and Discussion
N. equitans genome comprises a one chromosome of 490885 base pairs with GC content of 31.56%. This low GC content indicates that all the sense codons (61 codons) are not used uniformly, only the codons ending at third positions are A or T. In this study, 536 protein coding genes are recognized with an average base pair length of 848. Although N. equitans is smallest known Achaean, but protein coding and RNA genes of the genome are 95% of the total genome which is abnormally high in the genomes. Only 18.3% of the protein coding genes with unknown function are homologs to other genomes and residual genes are the sole features of this genome.20 In this work, MRCBS is applied to diverse genes from Nanoarchaeum equitans kin4-M and gene expression profiles are deduced by computing the MRCBS score for every gene and their distributions are shown in Figure 1. Figure 1 shows that distribution of the MRCBS score is unimodal and MRCBS score of most of the genes is varying from 0.633342 to 0.853183 and the mean and median MRCBS score are 0.766318 and 0.770313 respectively. The best threshold score of this genome is determined to 0.78. There are 186 predicted highly expressed genes (PHE) in Nanoarchaeum equitans Kin4-M by taking above mentioned threshold score. Table 1 shows the top ten genes as PHX gene. Functional class analysis [Annotation done by Secondary (JCVI) Annotation (http://cmr.jcvi.org/tigr-scripts/CMR/GenomePage.cgi)] are shown in Table 1. It is clear that out of 10 genes in top genes, 6 genes are ribosomal protein / translation factor which are commonly known as highly expressed genes.
 |
Figure 1: Distribution of MRCBS score for all protein coding genes in the Nanoarchaeum equitans kin4-M
|
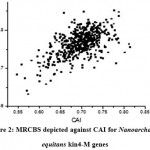 |
Figure 2: MRCBS depicted against CAI for Nanoarchaeum equitans kin4-M genes
|
Table 1: Top 10 MRCBS score of the genes in Nanoarchaeum equitans kin4-M genome
| Gene | Function | MRCBS |
| NEQ083 | 30S ribosomal protein S10 | 0.829214 |
| NEQ422 | diphthine synthase | 0.828606 |
| NEQ210 | prolyl-tRNA synthetase | 0.82274 |
| gltX | glutamyl-tRNA synthetase | 0.818104 |
| NEQ530 | 50S ribosomal protein L32e | 0.817878 |
| NEQ247 | 30S ribosomal protein S4 | 0.814004 |
| NEQ242 | 30S ribosomal protein S7 | 0.813881 |
| NEQ405 | translation initiation factor IF-2 alpha | 0.812336 |
| NEQ091 | 50S ribosomal protein L10 | 0.806075 |
| NEQ375 | 30S ribosomal protein S3a | 0.802264 |
The distinctive PHE genes in Nanoarchaeum equitans Kin4-M include rpoA2, rpoE1 and eef1b. Excluding 146 hypothetical genes, other PHE genes include RP genes ,protein synthesis, elongation factor EF-2 (NEQ543), , DNA-directed RNA polymerase subunit alpha(NEQ503), transcription antitermination protein NusG , exosome complex RNA-binding protein 1 (NEQ184), exosome complex exonuclease 2 (NEQ111), flap endonuclease-1 (NEQ088), N(2),N(2)-dimethylguanosine tRNA methyltransferase (NEQ108),glutamyl-tRNA synthetase gltX, phenylalanyl-tRNA synthetase beta subunit (NEQ479) ,prolyl-tRNA synthetase (NEQ210),proteasome-activating nucleotidase (NEQ186),threonyl-tRNA synthetase (NEQ177) , tyrosyl-tRNA synthetase (NEQ389),valyl-tRNA synthetase(NEQ252). Not all RP genes are PHE genes. Out of 36 RP genes, 18 RP genes are PHE genes. Excluding hypothetical genes top 20 PHE genes include 11 RP genes(S10,S4,S7,S3a,S17,S28e,S15,L32e,L10,L19e,L15), diphthine synthase(NEQ422), prolyl-tRNA synthetase (NEQ210), translation initiation factor IF-2 alpha subunit(NEQ405), tryptophanyl-tRNA synthetase(NEQ115),glutamyl-tRNA synthetase (gltX), leucyl-tRNA synthetase (leuS), DNA polymerase II small subunit ((NEQ240), DNA-directed RNA polymerase subunit E'(NEQ370), anaerobic ribonucleoside triphosphate reductase (NEQ345).
Table 2 shows the number of genes predicted versus total number of existing genes of this genome. It shows that majority of the genes present in the genome are highly expressed. Genes involved in functional class of biosynthesis of cofactors, prosthetic groups, and carriers, fatty acid and phospholipid metabolism, and Purines, pyrimidines, nucleosides, and nucleotides are not present in the genome, it implies that host genome is needed to synthesize amino acids and lipids. However, a percentage of genes (146 out of 444 hypothetical genes) characterized as hypothetical genes may be found among the PHE. Norman et al.21 showed that 238 out of 355 essential proteins in 16 model bacterial species comprised proteins in domains of unknown function (DUFs), and most of them represented single-domain proteins which obviously showed the biotic significance of DUFs. It suggests that experimental research should emphasis on conserved and essential DUFs for functional analysis. A detailed study of these putative/hypothetical PHE genes should yield a clear image of the replication and division machineries, and regulatory features of the cell cycle.
Table 2. No. of PHX genes predicted versus no. of genes present in Nanoarchaeum equitans kin4-M
| Functional Class | No. of Genes present in the genome | No. of highly expressed genes by MRCBS |
| Amino acid biosynthesis | 1 | 1 |
| Cell envelope | 1 | 1 |
| Cellular processes | 2 | 2 |
| Central intermediary metabolism | 1 | 1 |
| DNA metabolism | 8 | 7 |
| Energy metabolism | 2 | 2 |
| Protein fate | 5 | 4 |
| Regulatory functions | 2 | 2 |
| Transcription | 7 | 5 |
| Transport and binding proteins | 1 | 1 |
| Protein Synthesis | 59 | 58 |
Correlations Among Different Codon Bias Indices
In this study, expression patterns of 536 genes of Nanoarchaeum equitans kin4-M are obtained by computing the scores of CAI, RCA, RCBS and MRCBS. The predicted score MRCBS of a gene is computed by comparing its codon usage bias with the pro file of universally functional genes, such as the ribosomal protein (RP) genes. Using same ribosomal protein (RP) genes as reference set, used in the computation of MRCBS, CAI and RCA scores have been calculated. The results indicate that CAI is a strong correlated with RCA (r = 0.43644) and MRCBS (0.58787) while the correlation of RCBS with CAI is significantly worse (r = 0.0477). Figure 2 shows the comparison of the gene expression computed by MRCBS over CAI. It suggests that MRCBS can be used as a good estimator for codon usage bias.
Impact Codons
It is hypothesized that genes expressed at higher level uses a subset of optimal codons than the gene expressed at lower level. Impact codons propose such method to determine such codons in highly expressed genes by recognizing the level significance of z score. Thus, at the 95% level of significance, the following impact codons of Nanoarchaeum equitans genome are obtained: ttt, ttc (Phe), tta,ttg, cta (Leu), ata (Ile), tat, tac (Tyr), aaa (Lys), gaa, gag (Glu), tct, tcc (Ser), gcc, gct (Ala), tgg (Trp), cct, ccc, cca (Pro), agc, agg (Arg), ggt, ggc, ggg (Gly). Excluding Met, Val, Gln, Asp, Thr, Cys, Asn, His, it is observed that twenty five codons of 12 amino acids are found at higher significance level in PHE genes than the whole genome. Surprisingly, there is no compositional bias seen among these 25 codons which comprise 12 G/C and 13 A/T codons ending at third positions. So, all the impact codons are not GC rich and GC3 content could not be used as codon usage bias indicator at all. Thus, impact codons may possibly influence translational efficiency by selective forces acting on highly expressed genes.
Conclusion
Here, we analyzed the performance of MRCBS to forecast the expression profile of the Nanoarchaeum equitans kin4-M genome. Present study showed that most of the genes of this genome involved in amino acid biosynthesis, protein Synthesis, DNA metabolism and ribosomal / translation and transcription factor fall in PHE genes category. The other genes with PHE comprise cellular process, central intermediary metabolism, energy metabolism and many hypothetical proteins. In this study, good codon bias is observed in highly expressed genes of Nanoarchaeum equitans kin4-M genome. It can be noted that the performance of proposed methodology displays well correlated with expression measures computed by several other measures used in the literature. MRCBS relates codon bias in addition to the nucleotide biases of the genes for estimating the potential expression levels of genes. Due to biophysical and biochemical restrictions of the experimental methods, there are numerous limits to relate the connection between gene expression and codon bias. It has shown that the proposed methodology has performed well on a number of slow-growing and fast-growing bacteria genomes, Saccharomyces cerevisiae, Caenorhabditis elegans and Drosophila melanogaster,18 so MRCBS can possibly be used through relating it with other widely used codon usage measures.
Conflict of Interest
The author declares that he has no conflict of interest.
References
- Błażej P., Mackiewicz D., Wnętrzak M and Mackiewicz P. The Impact of Selection at the Amino Acid Level on the Usage of Synonymous Codons. G3 (Bethesda). 2017;7(3);967-981.
CrossRef - Mrázek J., Spormann A. M and Karlin S. Genomic Comparisons among γ-proteobacteria. Env. Microbiol. 2006;8:273-288.
CrossRef - Karlin S., Brocchieri L., Campbell A., Cyert M and Mrázek J. Genomic and proteomic comparisons between bacterial and archaeal genomes and related comparisons with the yeast and fly genomes. Proc. Natl. Acad. Sci. USA. 2005;102:7309-7314.
CrossRef - Karlin S., Mrazek J and Brocchieri M. L. Predicted highly expressed genes in archaeal genomes. Proc. Natl. Acad. Sci. USA. 2005;102:7303–7308.
CrossRef - Carbone A., Zinovyev A and Fékèps F. Codon Bias Signatures, Organization of Microorganisms in Codon Space and Lifestyle. Mol Biol Evol. 2005;22(3):547-561.
CrossRef - Zhou Z., Dang Y., Zhou M., Li L., Yu C., Fu J and Liu Y. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. USA. 2016;113(41):6117–6125.
CrossRef - Supek F and Vlahovicek K. Comparison of codon usage measures and their applicability in prediction of microbial gene expressivity. BMC Bioinformatics. 2005;6:182.
CrossRef - Supek F and Vlahovicek K. Correction Comparison of codon usage measures and their applicability in prediction of microbial gene expressivity. BMC Bioinformatics 2010;11:463.
CrossRef - Sharp P. M and Li W. H. The codon adaptation index -a measure of directional synonymous codon usage bias and its potential applications. Nucleic Acids Res. 1987;15:1281-1295.
CrossRef - Fox J. M and Erill I. Relative Codon Adaptation: A Generic Codon Bias Index for Prediction of Gene Expression. DNA Res. 2010;17:185–196.
CrossRef - Roymondal U., Das S and Sahoo S. Predicting gene expression level from relative codon usage bias an application to Escherichia coli genome. DNA Res. 2009;16:13–30.
CrossRef - Das S., Roymondal U and Sahoo S. Analyzing gene expression from relative codon usage bias in Yeast genome: a statistical significance and biological relevance. Gene. 2009;443:121– 131.
CrossRef - Das S., Roymondal U., Chottopadhyay B and Sahoo S. Gene expression profile of the cynobacterium synechocystis genome. Gene. 2012;497:344-352.
CrossRef - Kira S. M and Eugene V. K. Evolutionary and functional genomics of the Archaea. Current Opinion in Microbiology 2005;8(5):586-594.
CrossRef - Huber H., Christine M. Archaeal symbionts and parasites. Current Opinion in Microbiology. 2011;14(3):364-370.
CrossRef - Shields D. C. Switches in species-specific codon preferences: the influence of mutation biases. J. Mol. Evol. 1990;31:71–80.
CrossRef - Das S., Chakrabarti J., Ghosh Z., Sahoo S and Mallick B. A new measure to study phylogenetic relations in the brown algal order Ectocarpales: the codon impact parameter. J. Biosci. 2005;30:699–709.
CrossRef - Sahoo S and Das S. Analyzing gene expression and codon usage bias in diverse genomes using a variety of models. Current Bioinformatics. 2014;9:102-112.
CrossRef - Sahoo S and Das S. Analyzing gene expression and codon usage bias in Metallosphaera Sedula. Journal of Bioinformatics and Intelligent Control. 2014;3:72–80.
CrossRef - Bohlin J., Skjerve E and Ussery D. W. Analysis of genomic signatures in prokaryotes using multinomial regression and hierarchical clustering. BMC Genomics. 2009;10:487-496.
CrossRef - Norman F. G., Dietlind L. G., Peter U. Protein Domains of Unknown Function Are Essential in Bacteria. m.Bio. 2013;5(1):e00744-13.

This work is licensed under a Creative Commons Attribution 4.0 International License.



