

How to Cite | Publication History | PlumX Article Matrix
In Silico Structural Characterization of Plasmodium Falciparum Helicase, PfBrr2
Ritu Saxena and Prakash Chandra Mishra
Department of Biotechnology, Guru Nanak Dev University, Amritsar, Punjab, 143005, India.
Corresponding Author E-mail: pm.biotech@gndu.ac.in
DOI : http://dx.doi.org/10.13005/bbra/2657

ABSTRACT: Plasmodium falciparum is a causative agent of one of the most devastating disease, cerebral malaria. Absence of suitable vaccine and the emergence of multi drug resistant parasites hinder prevention of malaria disease worldwide. One of the most reliable approaches to control this disease is to develop antimalarial against drug targets which are specific for ubiquitous and necessary enzymes such as helicases. Helicases work in ATP dependent manner and help in unwinding of nucleic acids during replication, transcription and repair mechanism. In this study, in silico analysis and homology modeling method were used to characterize the physicochemical properties and 3D structure of PfBrr2 helicase. Suitable structure of different domains was validated using in silico tools and used for docking studies to understand protein-ligand interactions. Protein-protein interaction network of PfBrr2 was investigated to understand its function inside the parasite.
KEYWORDS: ATP; DEAD Box; Helicase; Sec63
Download this article as:| Copy the following to cite this article: Saxena R, Mishra P. C. In Silico Structural Characterization of Plasmodium Falciparum Helicase, PfBrr2. Biosci Biotech Res Asia 2018;15(3). |
| Copy the following to cite this URL: Saxena R, Mishra P. C. In Silico Structural Characterization of Plasmodium Falciparum Helicase, PfBrr2. Biosci Biotech Res Asia 2018;15(3). Available from: https://www.biotech-asia.org/?p=30947 |
Introduction
Plasmodium falciparum causes malaria which is transmitted to human by the bite of female anopheles mosquito (Akinosoglou et al, 2012). Five species of genus Plasmodium causes malaria in human, and these includes Plasmodium falciparum, Plasmodium ovale, Plasmodium malariae, Plasmodium knowelsi, Plasmodium vivax (Tuteja, 2007). Among these species Plasmodium falciparum poses great threat to human causing most severe form of this disease known as “cerebral malaria” (Cowman et al, 2016). According to WHO report on malaria in 2017, there was an estimated report of 445000 death globally, mostly in sub-Saharan African (Barber et al, 2017). Due to lack of any suitable vaccine, malaria control and prevention strategies include prevention of malaria transmission, early detection and prompt treatment of malaria cases (Cowman et al, 2016).
Malaria parasites are becoming resistant against various modern-day drug therapies (Ashley et al, 2014). Quinine is one of the first anti-malarial drug used for treatment followed by various other drugs available commercially such as Amodiaquine, Pyrimethamine, Proguanil, Sulfonamides, Mefloquine, Atovaquone, Primaquine. (Achan et al, 2011). Due to failure of these conventional drugs Artemisinin combination therapies (ACT) is being used, which includes Artemisinin with other drugs in combination. ACT’s have been highly effective but unfortunately reports of parasite’s resistance against Artemisinin in Southeast Asia further threaten the recent gains in malaria control and prevention (Duru et al, 2016 and Mishra et al, 2015).
Due to increasing resistance to modern day drug therapies, there is an urgent need to develop new antimalarials and to identify new drug targets which can combat with severity of disease (White, 1998). Various proteins essential for survival of the parasite such as helicases and protein translocation machinery (Sec 63) component of Plasmodium falciparum need to be explored as suitable antimalarial drug targets (Tuteja, 2007).
Helicases are universal enzymes present in bacteria, virus and eukaryotes including malaria parasite, Plasmodium (Tuteja and Pradhan, 2006). These principally acts on double stranded substrate which is designated as DNA-DNA, DNA-RNA, RNA-RNA depending on composition and catalyze the unwinding of substrate by utilising the energy which is provided by the intrinsic nucleic-acid dependent ATPase activity (Umate et al, 2011). Many cellular processes are driven by DNA helicases such as DNA replication, repair, recombination and transcription while RNA helicases play vital role in transcription, translation and RNA splicing (Tuteja 2003 and Cruz et al, 1999). Nine short conserved amino acid sequences called as helicase motifs are designated as Q, I, Ia, Ib, II, III, IV, V and VI in protein sequences of different species (Pradhan et al., 2007). Helicases are classified into six super families SF1 to SF6 based on their different signature motifs and these motifs are signature for “core domains” that form tandem RecA-like folds (Chauhan et al, 2017). Core domains include two universal features which include conserved residues involved in binding and hydrolysis of the NTP equivalent to the Walker A and B boxes of many ATPases; and an “Arginine finger” that plays a key role in energy coupling. Helicases are also classified as type A or B based on 3’-5’ or 5’-3’ translocation polarity. SF1, SF2 and SF6 contains both type and B helicases while SF3 are type A helicases and all members of SF4 and SF5 are type B (Chauhan et al, 2017).
In this study, we report in silico structural characterization of PF3D7_0422500, a homolog of pre-mRNA splicing helicase Brr2 where Brr stands for “Bad Response to Refrigeration”.
Tuteja in 2011 reported PfBrr2 as a homologue of ScBrr2p which has 32% sequence identity (Tuteja, 2011). However, PfBrr2 differs from ScBrr2p because it contains only one detectable helicase like domain while two domains are present in ScBrr2p (Tuteja, 2011). Recently, Zhang M et al used piggy Bac transposon insertional mutagenesis and quantitative insertion site sequencing to reach saturation level mutagenesis of parasite which leads into the identification of PfBrr2 as an essential gene for survival of parasite (Zhang et al, 2018). In silico approaches have been used to model the three-dimensional structure of different domains of PfBrr2. Validated structure was used for docking analysis with ATP and interactions were analysed using LigPLOT+. Protein-protein interaction network study was used to understand its function in parasite biology.
Materials and Methods
Amino Acid Sequence
Protein sequence of PF3D7_0422500 (PfBrr2) was retrieved from PlasmoDb (Bhal et al, 2003) for in silico analysis.
Physicochemical Characterization
Parameters like isoelectric point, extinction coefficient, aliphatic index, instability index and GRAVY were computed using ExPASy’s ProtParam web server (Gasteiger et al, 2005).
Domain Organization
Domain organization of protein sequence was analyzed by using Pfam (Bateman et al, 2004), InterPro (Apweiler et al, 2001) and motif analysis using PROSITE (Sigrist et al, 2009).
Molecular Modeling and Validation of Model
Blastp (Altschul et al, 1997) against RCSB protein databank (PDB) (Berman et al, 2006) was used to search for suitable templates for different domains of PfBrr2. Three-dimensional structures of different domains were predicted by SWISS-MODEL (Waterhouse et al, 2018) using corresponding templates. Predicted three-dimensional models were validated using ERRAT (Colovos and Yeates, 1993) and RAMPAGE (Lovell et al, 2003) for structural and stereochemical quality. Interactive visualization and analysis of molecular structures were done using PyMOL (Delano, 2016).
Molecular Docking
The docking analysis was carried out using the Patchdock v. beta 1.3 (Schneidman et al, 2005). SDF file of ATP was retrieved from Protein data bank (PDB) and converted to PDB file in UCSF chimera (Pettersen et al, 2004). The PDB files of PfBrr2 and ATP were uploaded in the Patchdock web-server with clustering RMSD value 1.5 and protein-small ligand type were selected as parameter for docking. Docked complex were visualized using Pymol and Ligplot+ (Wallace et al, 1995) was used to plot the interactions between protein and ATP.
Protein-Protein Interaction Network
STRING-DB version 10.5 (Szklarczyk et al, 2014) webserver was used for protein interaction network analysis of PfBrr2. Additionally, interacting partners from yeast two hybrid data from Plasmodb were also used for network analysis.
Results and Discussion
Physicochemical Characterization
Primary structure of protein determines the three-dimensional structure of a protein. In the present study, ExPASy’s Protparam tool was used to predict the physicochemical properties of protein (Table 1). The predicted molecular weight of PfBrr2 is 337.908 kDa while theoretical pI is 5.54. Aliphatic index is 89.62 which showed that relative volume of protein is occupied by aliphatic side chains. Negative Grand Average of hydropathicity of -0.674 indicates that the protein is hydrophilic and soluble in nature. Extinction coefficient was calculated to be 32769090 when all pairs of cysteine were assumed to form cysteines while when all cysteine were present in reduced form it is 325940.
Table 1: Showing physicochemical parameters.
| Parameter | Value |
| Molecular weight | 337.908 kDa |
| Theoretical pI | 5.54 |
| Aliphatic index | 89.62 |
| Grand Average of hydropathicity (GAVY) | -0.674 |
| Extinction coefficient (assuming all pairs of cys form cysteines) | 327690 |
| Extinction coefficient (assuming all pairs of cys are reduced) | 325940 |
Domain Organization
PfBrr2 is a large multidomain protein containing 2874 amino acid. Domain analysis reveals three important domains, DEAD box, C-terminal helicase and Sec63 Brl domain in PfBrr2. As per Pfam and Interpro results, DEAD/DEAH box helicase domain is present from 716 to 982 residues (Fig 1). The DEAD box is a subgroup of family SF2 and this family contains a conserved motif (Asp-Glu-Ala-Asp), DEAH box and Ski2-like domains. Partial genes of this family have been reported in P. falciparum and P. cynomolgi. All DEAD box proteins share ATP and nucleic acid binding sites, but these proteins have unrelated functions (Cordin et al, 2006). DEAD box helicase plays important role in ribosome biogenesis and acts in regulation of small ribosomal and nucleolar RNA’s (Cordin et al, 2006). In yeast there are various DEAD box proteins which play important role in RNA splicing includes Prp2p, Prp5p, Prp16p, Prp28p, Prp43p and Brr2. Plasmodium falciparum also harbors homology of these helicases except Prp22p. These helicases perform different functions such as Prp5 and Prp28p function early in spliceosome assembly whereas Brr2p functions are needed both during assembly and disassembly of spliceosome (Singh et al, 2012).
Sec63 domains are predicted by Pfam, and Interpro in PfBrr2 from 1379 to 1706 and 2503 to 2869 residues (Fig 1). Sec63 domain (also known as Brl domain) is present in proteins involved in endoplasmic reticulum translocons, pre-mRNA splicing helicase BRR2, HFM1 protein and putative helicases (Tuteja, 2007). Brl domain which exhibits homology to yeast U5 RNA helicase is required for the formation of SEC complex (Tormyet al, 2006). Furthermore, Brl domain acts as protein binding domain within Sec63p during complex assembly. Thus, it can be speculated that, as in spliceosome, it might also help in structural remodeling events within translocon during this complex reaction process (Tormy et al, 2006).
ATP binding site is predicted to be present within the DEAD/DEAH box helicase from residue 738-747 while from 850-853 residues Magnesium binding site were predicted. A helicase conserved C-terminal domain from 1023 to 1181 residues is predicted using Pfam, Interpro and PROSITE (Fig 1).
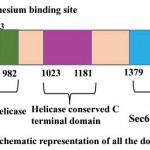 |
Figure 1: Schematic representation of all the domains present in PfBrr2.
|
Three-Dimensional Modelling and Validation
Three-dimensional structure of protein helps in prediction of the function of protein and also in rationale structure-based drug designing. Templates for homology modeling were selected using Blastp result against PDB database based on the sequence identity, query coverage and function of the protein (Table 2). For each domain different templates were selected which include S. cerevisiae spliceosomal helicase BRR2 (PDB ID: 5M5P) for DEAD/DEAH box helicase, crystal structure of Chaetomium thermophilum Brr2 helicase core (PDB ID: 5M59) for helicase C terminal domain, U5 small nuclear ribonucleoprotein 200 kDa helicase (PDB ID: 5URJ) for Sec63 Brl I domain and Brr2 Helicase Region S1087L (PDB ID: 4F92) for Sec63 Brl II domain. Template sequence were aligned with the sequence of the respective domains of PfBrr2 (Fig 2). Four structures were predicted by SWISS-MODEL (Fig 3) and these were evaluated using ERRAT and RAMPAGE (Fig 4 and Fig 5). The score of best structure of DEAD box helicase was 93.6% and 85.9 for Rampage and ERRAT respectively. In case of Helicase C terminal conserved region 95.5% residues fall under favourable region in Rampage and good quality factor, ERRAT was found to be 86.25. Sec Brl domain I and II have 95.5% and 95.1% residues respectively in favorable region as per Rampage results. ERRAT was 84.85 and 86.85 for Brl I and II domain respectively. ERRAT and Rampage validated scores suggest that all predicted 3D models are reliable and can be used further for study. Tabular presentation of scores of Rampage and ERRAT for predicted models are given in table 3.
Table 2: Templates used for modeling PF3D7_0422500 with their PDB’ ids, percentage of query cover and percent identity.
| Source organism and PDB ID | Domain of protein | Residue number | Percent identity |
| S. cerevisiae spliceosomal helicase BRR2 (271-end) in complex with the 2 JAB/MPN domains of S. Cerevisiae PRP8 (PBD ID: 5M5P) | DEAD/DEAH box helicase | 715-982 | 53.50% |
| Crystal structure of Chaetomium thermophilum Brr2 helicase core in complex with Prp8 Jab1 domain (PDB ID: 5M59) | Helicase conserved C terminal domain | 1022-1182 | 58.60% |
| U5 small nuclear ribonucleoprotein 200 kDa helicase (PDB ID: 5URJ) | Sec63 Brl domain I | 1378-1706 | 45.21% |
| Brr2 Helicase Region S1087L (PDB ID: 4F92) | Sec63 Brl domain II | 2502-2867 | 34.52% |
Table 3: ERRAT and RAMPAGE scores of best models for different domains.
| Domain | ERRAT | RAMPAGE |
| DEAD/DEAH box helicase | 85.4 | 93.6% |
| Helicase conserved C terminal domain | 86.25 | 95.5% |
| Sec63 Brl domain I | 84.85 | 95.5% |
| Sec63 Brl domain II | 86.85 | 95.1% |
Structural analysis shows that DEAD/DEAH box helicase domain of PfBrr2 comprises of six alpha helices and four beta sheets connected by loops. Predicted ATP binding site are formed by beta sheets while few residues fall both in loops and alpha helix. Helicase C terminal domain consists of six alpha helices and five beta sheets. C-terminal of PfBrr2 contains two Sec63 Brl domains and each consists of various alpha helices and beta sheets. Story and Steitz (1992) reported crystal structures of SF1 and SF2 helicases which contain two covalently linked globular domains having five beta strands surrounded by five alpha helices (Story and Steitz, 1992). In our study predicted structures of different domains of PfBrr2 consists of similar domain containing five alpha sheets and five beta strands. However, in our study we are not able to predict the structure of full length protein due to lack of suitable templates, but the predicted structure of each domain contains same number of alpha helices and beta sheets. This showed that modeled structure is suitable for further structural analysis.
 |
Figure 2: Sequence alignment of query and template sequence: A. DEAD box helicase domain. B. helicase conserved C terminal helicase domain. C. Sec63 Brl domain I D. Sec63 Brl domain II.
|
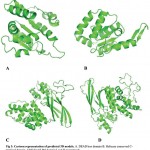 |
Figure 3: Cartoon representation of predicted 3D models. A. DEAD box domain B. Helicase conserved C-terminal domain. C&D Sec63 Brl domain I and II respectively.
|
 |
Figure 4: Representation of Ramachandran plots of different modelled domains of PfBrr2 A. DEAD box domain. B. Helicase conserved C-terminal domain. C&D Sec63 Brl domain I and II respectively.
|
 |
Figure 5: ERRAT score validation of 3D structures of different domains of PfBrr2 A. DEAD box domain. B. Helicase conserved C-terminal domain. C&D Sec63 Brl domain I and II respectively.
|
Analysis of ATP Binding Site
ATP binding sites were predicted using conserved domain database (CDD) and PROSITE. Binding of ATP is a crucial step in the function of helicases as ATP binding is required for strand separation. Docking studies were used to understand the binding of ATP with three-dimensional structure of PfBrr2. ATP was present in the docked complex of Patchdock at a similar as site predicted using CDD database. Residues involved in binding with ATP are Methionine-734, Isoleucine-736, Proline-741, Threonine-742, Glycine-743 and Lysine-746 as per Ligplot analysis of complex structure (Fig 6A & B). Distance between different interacting atoms of ATP and interacting residues were measured using PyMOL (Fig 7). O3 atom of ATP interacts with carbonyl oxygen of methionine while its O1 atom interacts with carbonyl oxygen of phenylalanine. O7 atom interacts with N of glycine while O10 and O11 interacts with nitrogen of lysine. ATP is observed to be completely embedded in the pocket of protein structure (Fig 8).
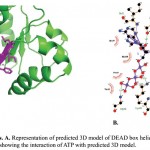 |
Figure 6: Docking analysis. A. Representation of predicted 3D model of DEAD box helicase domain with docked ATP moiety. B. Ligplot showing the interaction of ATP with predicted 3D model.
|
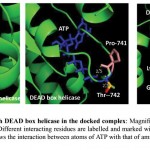 |
Figure 7: Interaction of ATP with DEAD box helicase in the docked complex.
|
Magnified view of interacting surface of DEAD box helicase and ATP. Different interacting residues are labelled and marked with different colours while ATP is in blue colour. Dotted lines shows the interaction between atoms of ATP with that of amino acid and distance is measured in angstrom.
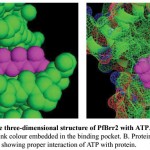 |
Figure 8: Representation of the three-dimensional structure of PfBrr2 with ATP.
|
A. Docked complex is shown in sphere form and ATP in pink colour embedded in the binding pocket. B. Protein is shown as mesh form while ATP in sphere in pink colour showing proper interaction of ATP with protein.
Potential Interacting Partners
In silico analysis for interacting partners of PfBrr2 was done using STRING-DB and yeast two hybrid data from Plasmodb. STRING-DB results show that PfBrr2 interacts with various proteins which play important role in RNA splicing and maturation (Fig 9A & B). During gene expression, genes are expressed as precursor mRNA (pre-mRNA) and are converted to mRNA by splicing. During splicing non-coding sequences of introns are removed while coding sequences of exons are ligated. Nuclear pre-mRNA splicing is catalyzed by spliceosome, a multi-megadalton ribonucleoprotein (RNP). Each spliceosome is composed of five snRNAs named U1, U2, U4, U5, U6 and range of associated protein factors. Result from STRING database reveals that PfBrr2 interacts with various proteins homolog present in the spliceosome complex including RNA helicases (Pf10_0294 and PF08_0042), pre-mRNA splicing factor (PFD0265w and PFL0970w) and components of spliceosome U5 and U2 (PF11_0108, PFC037c and PF10_0041) (Fig 9C & Table 4). In case of yeast and humans, many proteins such as Prp11, Prp9, Prp5, Prp16, Prp17, Prp21 interact with U2 snRNP’s to form complex and this complex helps in pre-mRNA splicing (Ruby et al, 1993 and Zhou and Reed, 1998). Additionally, yeast two-hybrid data from Plasmodb reveals that three more proteins of Pf interact with PfBrr2 and these includes adenosyl homocysteinase, metallohydrolase/oxidoreductase and ABC transporter of family1 (Table 5). Adenosyl homocysteinase is competitive inhibitor of S-adenosyl-L-methionine dependent methyl transferase hence plays key role in regulation of intracellular concentration of adenosyl homocysteine while metallohydrolase plays role in redox reactions (Turner et al, 2000 and Hai, 2016). ABC transporter are transmembrane proteins, and these helps in transport of ligands across biological membranes (Linton, 2007). Protein-protein interaction study reveals the role of PfBrr2 in RNA splicing and its presence as one of the components of spliceosome which may be formed by various interacting proteins.
 |
Figure 9: Protein-protein interaction: A. The results of STRINGS-DB database show the proteins interacting with PrBrr2p. The plasmodb ID’s of all are shown in the model. B. Explanation of interactions shown in A. C. A model of different interacting partners of PfBrr2p.
|
Table 4: Interacting partners of PfBrr2 predicted from STRING-DB database.
| Plasmodb ID | Name of interacting protein | Part of spliceosome | Score |
| PF10_0294 | RNA helicase putative (1290 aa) | Yes | 0.978 |
| PFD0265w | Pre-mRNA splicing factor, putative (3136 aa) | Yes | 0.968 |
| PF11_0108 | U5 snRNP associated protein, putative (1235 aa) | Yes | 0.926 |
| PFC0375c | U2 snRNP spliceosome subunit, putative (1386 aa) | Yes | 0.892 |
| PFL1735c | RNA processing protein, putative (1031 aa) | Yes | 0.875 |
| PF10_0041 | U5 small nuclear ribonuclear protein, putative (1235 aa) | Yes | 0.859 |
| PFC0365w | PRP19-like protein, putative (532 aa) | No | 0.852 |
| PFL0970w | Pre-mRNA splicing factor, putative (618 aa) | Yes | 0.848 |
| PFL1855w | Cell-cycle control protein, putative (967 aa) | No | 0.830 |
| PF08_0042 | ATP dependent RNA helicase prh1, putative (867 aa) | Yes | 0.828 |
Table 5: Interacting partners from Yeast two hybrid data:
| Gene | Interacts with | Name of interacting protein | Bait start | Bait end | Prey start | Prey end |
| PF3D7_0422500 | PF3D7_0520900 | adenosyl homocysteinase | 26 | 296 | 6326 | 6655 |
| PF3D7_0422500 | PF3D7_0723700 | Metallohydrolase/oxidoreductase, putative | 671 | 755 | 2133 | 2590 |
| PF3D7_0422500 | PF3D7_0813700 | ABC transporter F family member 1, putative | 370 | 431 | 6493 | 6659 |
Conclusion
Helicases are vital class of enzymatic tools which are necessary for various nucleic acids metabolic pathways. These are also considered as “screw driver” of cellular machinery and study of these molecules would help to understand the malaria parasite biology. Additionally, Zang et al in 2018 reported PfBrr2 as one of the essential proteins for survival of malaria parasite inside host cell. In silico domain analysis reveals three domains in PfBrr2 protein including DEAD box helicase, helicase conserved C terminal domain and Sec63 Brl domain I and II. Three-dimensional predicted structure of DEAD box helicase domain contains six alpha helices and four beta sheets, helicase conserved C terminal domain consists of six alpha helices and five beta sheets while Sec63 Brl domain I harbor nine alpha helices and seven beta sheets whereas ten alpha helices and seven beta sheets are present in Sec Brl domain II. Docking studies were used to study the residues and interactions involved in the binding pocket of ATP. Protein-protein interaction analysis reveals that it is present in spliceosomal complex and plays an important role in splicing and maturation of pre-mRNA. Three-dimensional structure of this protein can be explored for development of antimalarials.
Acknowledgements
Ritu Saxena is a Department of Biotechnology-Senior Research Fellow (DBT-SRF), Govt. of India.
Conflict of Interest
Authors declare no conflict of interest.
References
- Akinosoglou K. S., Solomou E. E & Gogos C. A. Malaria a haematological disease. Hematology. 2012;17(2):106-114.
CrossRef - Tuteja R. Malaria− an overview. The FEBS journal. 2007;274(18):4670-4679.
CrossRef - Cowman A. F., Healer J., Marapana D & Marsh K. Malaria biology and disease. Cell. 2016;167(3):610-624.
CrossRef - Barber B. E., Rajahram G. S., Grigg M. J., William T & Anstey N. M. World Malaria Report time to acknowledge Plasmodium knowlesi malaria. Malaria journal. 2017;16(1):135.
CrossRef - Ashley E. A., Dhorda M., Fairhurst R. M., Amaratunga C., Lim P., Suon S & Sopha C. Spread of artemisinin resistance in Plasmodium falciparum malaria. New England Journal of Medicine. 2014;371(5):411-423.
CrossRef - Achan J., Talisuna A. O., Erhart A., Yeka A., Tibenderana J. K., Baliraine F. N & D’Alessandro U. Quinine an old anti-malarial drug in a modern world: role in the treatment of malaria. Malaria journal. 2011;10(1): 144.
CrossRef - Duru V., Witkowski B & Ménard D. Plasmodium falciparum resistance to artemisinin derivatives and piperaquine: a major challenge for malaria elimination in Cambodia. The American journal of tropical medicine and hygiene. 2016;95(6):1228-1238.
CrossRef - Mishra N., Prajapati S. K., Kaitholia K., Bharti R. S., Srivastava B., Phookan S & White N. J. Surveillance of artemisinin resistance in Plasmodium falciparum in India using the kelch 13 molecular marker. Antimicrobial agents and chemotherapy. 2015;59(5):2548-2553.
CrossRef - White N. J. Drug resistance in malaria. British Medical Bulletin. 1998;54(3):703-715.
CrossRef - Tuteja R. Helicases− feasible antimalarial drug target for Plasmodium falciparum. The FEBS journal. 2007;274(18):4699-4704.
CrossRef - Tuteja R & Pradhan A. Unraveling the ‘DEAD-box’helicases of Plasmodium falciparum. Gene. 2006;376(1):1-12.
CrossRef - Umate P., Tuteja N &Tuteja R. Genome-wide comprehensive analysis of human helicases. Communicative & integrative biology. 2011;4(1):118-137.
CrossRef - Tuteja N. Plant DNA helicases: the long unwinding road. Journal of experimental botany. 2003;54(391):2201-2214.
CrossRef - la D., Cruz J., Kressler D & Linder P. Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends in biochemical sciences. 1999;24(5):192-198.
CrossRef - Pradhan A., Chauhan V. S & Tuteja R. DEAD-box ‘he licase from Plasmodium falciparum is active at wide pH and is schizont stage-specific. Journal of vector borne diseases. 2007;44(1):12.
- Chauhan M., Tarique M &Tuteja R. Plasmodium falciparum specific helicase 3 is nucleocytoplasmic protein and unwinds DNA duplex in 3′ to 5′ direction. Scientific Reports. 2017;7(1):13146.
CrossRef - Tuteja R. Helicases involved in splicing from malaria parasite Plasmodium falciparum. Parasitology international. 2011;60(4):335-340.
CrossRef - Zhang M., Wang C., Otto T. D., Oberstaller J., Liao X., Adapa S. R & Brown J. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science. 2018;360(6388):7847.
CrossRef - Bahl A., Brunk B., Crabtree J., Fraunholz M. J., Gajria B., Grant G. R & Li L. PlasmoDB the Plasmodium genome resource. A database integrating experimental and computational data. Nucleic acids research. 2003;31(1):212-215.
CrossRef - Gasteiger E., Hoogland C., Gattiker A., Wilkins M. R., Appel R. D & Bairoch A. Protein identification and analysis tools on the ExPASy server. In The proteomics protocols handbook. 200;571-607. Humana press.
- Bateman A., Coin L., Durbin R., Finn R. D., Hollich V., Griffiths‐Jones S & Studholme D. J. The Pfam protein families database. Nucleic acids research. 2004;32(1):138-141.
CrossRef - Apweiler R., Attwood T. K., Bairoch A., Bateman A., Birney E., Biswas M & Durbin R. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic acids research. 2001;29(1):37-40.
CrossRef - Sigrist C. J., Cerutti L., Castro D .E., Langendijk-Genevaux P. S., Bulliard V., Bairoch A & Hulo N. PROSITE a protein domain database for functional characterization and annotation. Nucleic acids research. 2009;38(1):161-166.
- Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W & Lipman D. J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research. 1997;25(17):3389-3402.
CrossRef - Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H & Bourne P. E. The protein data bank, 1999–. In International Tables for Crystallography Volume F. Crystallography of biological macromolecules. 2006;675-684. Springer Netherlands.
- Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R & Lepore R. SWISS-MODEL homology modelling of protein structures and complexes. Nucleic acids research. 2018.
CrossRef - Colovos C., & Yeates T. O. Verification of protein structures patterns of no nbonded atomic interactions. Protein science. 1993;2(9):1511-1519.
CrossRef - Lovell S. C., Davis I. W., Arendall W. B., Bakker D. P. I., Word J. M., Prisant M. G & Richardson D. C. Structure validation by Cα geometry and Cβ deviation. Proteins Structure, Function and Bioinformatics. 2003;50(3):437-450.
CrossRef - Lano D. W. L. The Py M O L Molecular Graphics System. De Lano Scientific Palo Alto, CA: 2002. There is no corresponding record for this reference. 2016.
- Schneidman-Duhovny D., Inbar Y., Nussinov R & Wolfson H. J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic acids research. 2005;33(2):363-367.
CrossRef - Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C & Ferrin T. E. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25(13):1605-1612.
CrossRef - Wallace A. C., Laskowski R. A & Thornton J. M. LIGPLOT a program to generate schematic diagrams of protein-lig and interactions. Protein engineering, design and selection. 1995;8(2):127-134.
CrossRef - Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J & Kuhn M. STRING v 10 protein–protein interaction networks, integrated over the tree of life. Nucleic acids research. 2014;43(1):447-452.
- Cordin O., Banroques J., Tanner N. K & Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17-37.
CrossRef - Singh P. K., Kanodia S., Dandin C. J., Vijayraghavan U & Malhotra P. Plasmodium falciparum Prp 16 homologue and its role in splicing. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms. 2012;1819(11):1186-1199.
CrossRef - Tuteja R. Unraveling the components of protein translocation pathway in human malaria parasite Plasmodium falciparum. Archives of biochemistry and biophysics. 2007;467(2):249-260.
CrossRef - Jermy A. J., Willer M., Davis E., Wilkinson B. M & Stirling C. J. The Brl domain in Sec 63 p is required for assembly of functional end oplasmic reticulum translo cons. Journal of Biological Chemistry. 2006;281(12):7899-7906..
CrossRef - Story R. M & Seitz T. A. Structure of the recA protein–ADP complex. Nature. 1992;355(6358):374.
CrossRef - Ruby S. W., Chang T. H & Abelson J. Four yeast spliceosomal proteins (PRP 5, PRP 9, PRP 11 and PRP 21) interact to promote U2 snRNP binding to pre-mRNA. Genes & development. 1993;7(10):1909-1925.
CrossRef - Zhou Z & Reed R. Human homologs of yeast Prp 16 and Prp 17 reveal conservation of the mechanism for catalytic step II of pre‐mRNA splicing. The EMBO Journal. 1998;17(7):2095-2106.
CrossRef - Turner M. A., Yang X., Yin D., Kuczera K., Borchardt R. T & Howell P. L. Structure and function of S-adenosylhomocysteine hydrolase. Cell biochemistry and biophysics. 2000;33(2):101-125.
CrossRef - Hai Y. Structure and function of metallohydrolases in the arginasedeacetylase family. 2016.
- Linton K. J. Structure and function of ABC transporters. Physiology. 2007;22(2):122-130.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.



