

How to Cite | Publication History | PlumX Article Matrix
Homology Modeling of Bifunctional Enzyme Alanine Racemase from Taibaiella Chishuiensis
Gulsanga lemar and Farid Ahmad Danish Far
Biosciences, Faculty of Science, University Teknologi Malaysia, Malaysia.
DOI : http://dx.doi.org/10.13005/bbra/2864

ABSTRACT: Alanine Racemase from Taibaiella chishuiensis bacteria is one of the bifunctional enzymes that catalyze the L- and D-alanine racemization of peptidoglycan biosynthesis in bacteria and ligation (UDP-N-acetylmuramoyl-Tripeptide-D-alanyl-D-alanine ligase). It had two EC numbers 5.1.1.1 and 6.3.2.10 respectively. This enzyme is an important target for antimicrobial drug productions or inhibitor design. However, the 3D structure of Alanine Racemase from Taibaiella or UDP-N-acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase/alanine racemase has remained unknown. Thus, this study modeled and validated the 3D structure of the enzyme in the query. The bioinformatics tools/databases and software such as BRENDA, NCBI, UniProt, Clustal Omega, ProtParam, Swiss model, Phyre2, GOR, PROCHECK, and PyMOL were used for modeling, validation, and structural comparison. From the sequence and 3D structure analysis, it is indicated that Alanine racemase from Taibaiella had the same active and binding sites with the reference enzymes. Thus, we were able to study the similarities and differences in the sequence and structural properties of alanine racemase in two different bacteria. Finally, it was found that our enzyme has two parts for two different functions (racemization and ligation). The predicted model of alanine racemase of T. chishuiensis from this study could serve as a useful model for further study regarding the other bifunctional enzymes structure and function as well as drug design projects.
KEYWORDS: Active Site; Alanine Racemase; Bifunctional Enzyme; Binding Site; Bioinformatics Tools; Predicted Model; Taibaiella chishuiensis;
Download this article as:| Copy the following to cite this article: Lemar G, Far F. A. D. Homology Modeling of Bifunctional Enzyme Alanine Racemase from Taibaiella Chishuiensis. Biosci Biotech Res Asia 2020;17(3). |
| Copy the following to cite this URL: Lemar G, Far F. A. D. Homology Modeling of Bifunctional Enzyme Alanine Racemase from Taibaiella Chishuiensis. Biosci Biotech Res Asia 2020;17(3). Available from: https://bit.ly/3dtYKR1 |
Introduction
Alanine racemase, which belongs to the isomerases family, is a bacterial enzyme with EnzymeClassification (EC) number: 5.1.1.1(Muhammad and Zhao, 2019). It catalyzes the racemization of L- and D- alanine(Figure1),and pyridoxal5’- phosphate (PLP) is required as a cofactor (Muhammad et al.,2019). Woodand Gunsalus discovered and isolated this enzyme in Streptococcus faecalis, for the first time (Wood and Gunsalus, 1959).
This enzyme has a significant role in the growth of the bacteria by providing D-alanine, the peptidoglycan layer constitutive of the bacterial cell wall. (Liu et al., 2018). In both gram-positive and Gram-negative bacteria, the peptidoglycan layer of the bacterial cell wall prepares resistivity to osmotic lysis (Islam etal., 2017). Alanine racemase is special to bacteria. There are few exceptions to the fact that certain eukaryotes have the enzyme D- alanine-containing peptides biosynthesis in fungi, D- alanine metabolism in yeast, as well as osmotic regulation in a crayfishplusa bivalve mollusk (Nomura et al., 2001).
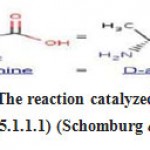 |
Figure 1: The reaction catalyzed by alanine racemase (5.1.1.1) (Schomburg et al., 2004). |
This enzyme is absent in humans and is a unique enzyme that transforms L-alanineandD- alanine in most bacteria thus, it is an important target for antimicrobial drug productions(Im et al., 2011; Wei et al., 2016). For instance, alanine racemase was chosen as a target to discover new drugs to treat tuberculosis or for the inhibitor design (Anthony et al., 2011). Based on previous studies, most of previously studied inhibitors suffer from the lack of specificity, and further investigation is required to solve this issue(Azam and Jayaram, 2016). To target an enzyme for effective antibacterial drugs, and to develop new and selective inhibitors, which improves treatment in public health systems, it is important to know about the structure and characteristics of the enzyme in various living organisms. The 3D structure of the alanine racemase of several bacteria is available in the PDB. But there is no 3DstructureoftheTaibaiella chishuiensis alanine racemase. Strain AY17 T of Taibaiella chishuiensis contained MK-7 as the predominant respiratory quinone, plus the main polar lipid phosphatidylethanolamine, and hydrolyses the aesculin, casein, and gelatin (Tan et al., 2014). Therefore, in the current study, we perform modelling of alanine racemase in the mentioned bacteria. The present investigation is the first study of the sequence and 3D structural characterization of the Taibaiella chishuiensis alanine racemase enzyme.This study modeled the structure of the novel bifunctional, Taibaiella chishuiens-based alanine racemase, which belongs to the family of isomerases. A bacterial strain, AY17 T of Taibaiella chishuiensis, was isolated from the Chishui River in Guizhou Province, SouthwestChina.
Based on phenotypic, phylogenetic, plus genetic evidence,theAY17Tstrain was categorized as a novel representative species of the Taibaiella genus for which theTaibaiella chishuiensissp.Nov., the name has been suggested. This bacteria belongs to the Chitinophagaceae family (Wei et al., 2016). The structure of the alanine racemase was first performed on an enzyme isolated from Bacillus stearothermophilus. This enzyme is a homodimeric enzyme with each monomer consisted of α/β barrel domain at the N-terminus and a C-terminal domain consisting mainly of β strands. The location of the active site is at the interface of the α/β barrel andthe β domain, close to the PLP cofactor, forming an internal aldimine connection to the lysine residue (Im et al., 2011). Crystal structure study has shown that almost all alanine racemases are dimeric (Dong et al., 2018). Mutagenic, modeling and structural analyzes indicate Tyr265 and Lys39 of G.Stearothermophilus alanine racemase are residues involved in the movement of protons to the racemase reaction. Lys39 and Tyr265 residues are composed of two similar polypeptides. These two residues are well preserved in other Alanine racemases (Ju etal., 2011).
To clarify the bifunction potency of the alanine racemase enzyme, a comparative study was performed between the primary sequence and the predicted structure of the new bifunctional alanine racemase enzyme and similar, clostridium difficile (PDB: 4LUT) from the family, Peptostreptococcaceaeas as well as T. maritime (PDB: 3zl8) from the family, thermotogaceae (Couñago et al., 2009; Tan et al., 2014; Dong et al., 2018).
Finally, several analyses were performed to provide useful details on the bifunctionality of this enzyme. This study wishes to provide useful information for modeling/designing other unknown structures of the bifunctional enzymes as well as drug design projects.
Research Methods
Sequence Retrieval and Multiple Sequence Alignment(MSA)
The information about the enzyme alanine Racemase of T. chishuiensis is obtained by searching the EC number (5.1.1.1) in BRENDA (Schomburg et al., 2004). So, we found out the optimum temperature,PH,metabolic path way reaction.Also, the FASTA format and other information regarding the PDB structure is received from UNIPROT (Ju et al., 2011). Then, the Blastp from NCBI database was done to obtain a similar sequence of alanine racemase from different organism to compare and analyze the structure (Asojo et al., 2014). Based on the data, it was found that our enzyme is very similar to bifunctional enzymes “UDP-N- acetylmuramoyl-tripeptide-D-alanyl-Dalanine ligase/alanine racemase” of different bacteria especially genus Taibaiella and Chitinophagaceae bacterium. But all of them do not have a PDB structure (Chen et al.,2019).
The primary structure analysis model of enzyme alanine racemase from T. chishuiensis was done using the ProtParam tool (Gasteiger et al., 2003;Hassanetal.,2020).This step was carried out to identify the estimated molecular weight, the theoretical pI value, the composition of the amino acid, the total amount of positively charged plus negatively charged residues, the atomic composition, the chemical formula, the aliphatic index, and the hydropathicity value of the model protein.
Secondary Structure Prediction Plus 3D Structure-Modeling
The secondary structure of enzyme alanine racemase in Taibaiella chishuiensis was predicted using the GOR database (Hassan et al., 2020). The 3D structure of the alanine racemase of T. chishuiensis was modeled using the SWISS- MODEL (Biasini et al., 2014). The best model generated from SWISS-MODEL was selected on the basis of a high sequence identity score. Our enzyme (alanine racemase of Taibaiella chishuiensis) was similar to two different enzymes: UDP-N-acetylmuramoyl-tripeptide-Dalanyl-D- alanine ligase (PDB ID: 3zl8) plus monomer of alanine racemase (PDB ID: 4lut). Also, PHYRE2 modeler was used for our sequence to build the 3D model (Kelley et al.,2015).
Homology Model Validation
The predicted 3D structure was validated using the PROCHECK software and Ramachandran plot to investigate the psi-phi angles to determine the accuracy of the predicted structure. Verify 3D is also used to evaluate the 3D protein structure model (Meo and Cozzetto, 2006).
Structural Comparison
The structure of alanine racemasefrom chishuiensis was analyzed and compared with alanine racemase from C. difficile.PyMol was used to superimpose the modeled structure of the alanine racemase of T. chishuiensis and the known structure of the alanine racemase of difficile. Since our enzyme is bifunctional, act also as a ligase, thus, we compared with the known structure of UDPN-acetylmuramoyl- tripeptide-D-alanyl-D-alanine ligase (PDB ID: 3zl8). For comparison of the surface structure and determining active site and binding site, we used also the PyMol (Meo and Cozzetto,2006)
Result and Discussion
Primary Sequence
The amino acids sequence (FASTA format) of alanine racemase of T. chishuiensis retrieved from the UniProt database (Consortium, 2015).>tr|A0A2P8D088|A0A2P8D088_9BACT Alanine racemase OS=Taibaiella chishuiensis OX=1434707 GN=B0I18_10740 PE=3 SV=1 MNLNDILKATKAEAIPEVKDIDIAELLIDSR KIVNPARAMFIAIKGSHRDGHQFIDDAYEK GVRTFLISEETDTAKYKDAQFLKVTDTLTA LHQLATAHRNQFHIPVIGITGSNGKTIVKE WLFQLLSENYKIIRSPKSYNSQIGVPLSVWR MQAHHTLGIFEAGISQPGEMSRSERILHPDI GIFTNIGEAHSEGFLNNRQKINEKLILFRHA KELIYCKDHHELNECIVQYCHQIRSGEEDD LKVFTWSQKSEATLRIQGIDKSSNSTTIRGV YEEREIAILIPFSDNASIENAIHCWCVLLLFK IEDSQIAERMARLHPVAMRLELLHGVDNCT IINDTYNSDLTSLQIALDFLEQQKQHPRRTV ILSDMLQIGKPDGDLYEEVAELVSQKNIYRI ICIGVALSKHKPTFRKHKKLRSIFFKSTEEFL KKMHMITFENEAILLKGARAFRFEKIEKLL EQKIHKTVLEINLSAIQNNLNVYRALLPAPV KMMAMVKAYSYGSGSYEIANLLQFAGVD YLTVAYVDEGVALRKAGISAPIMVMSPEA GAFDRMISWKLEPEIFSIYSLQQFTEIAHTL NVSHYPVHIKLDTGMHRLGFMPHELPELM AELKVNTAIKVVSIFSHLAGSDSADFDGFT AEQARNFDNMSQELMQVLPTRPLRHLVNS TGITRHPELYYDMVRLGIGMYGFDGSDKV QHQLQNIGTLKTTIAQIKELKAGETVGYSR KGKLERDSRIATVSIGYADGYFRDFGNGNA HMLVHGKPAPVVGAVCMDMCMLDITGID NAAVGDEVIVFGNELPIGQLADWAKTIPYE VLTGISQRVNRIYVNE
Primary structure analysis of the alanine racemase sequence was done using of Protpram tool. The protein informational analysis is in table 1.
Table 1: Calculation of some physical and chemical parameters of alanine racemase from T. chishuiensis (Gasteiger et al., 2003).
| The amino
acids number |
831 |
| Molecular
weight |
93816.05 |
| Theoretical pI | 6.42 |
| Total amount of positively charged residues
(Arg + Lys) |
92 |
| Total amount of negatively charged residues
(Asp + Glu) |
101 |
|
Atomic composition |
Hydrogen H 6656
Carbon C 4197 Oxygen O 1222 Nitrogen N 1144 Sulfur S 35 |
| Formula | Formula:
C4197H6656N1144O1222S35 |
| Total amount
of atoms |
13254 |
|
The estimated half-life |
30 hours (mammalian reticulocytes, in vitro)
>10 hours (Escherichia coli, in vivo) >20 hours (yeast, in vivo) |
| Aliphatic
index |
97.30 |
| Grand average of
hydropathicity (GRAVY) |
-0.160 |
Multiple Sequence Alignment
From the BLAST results obtained from NCBI, alanine racemase was chosen from the five different species to perform the multiple sequences alignment (Table 2). These 5 enzymes were aligned by using Clustal Omega which is shown in Figure 2. Positions with a single, completely conserved residue have been demonstrated by an asterisk (*), conservation between groups with highly similar characteristics were indicated by a colon (:), conservation between groups with less similar characteristics were indicated by a period (.) and the variable regions were indicated by a gap in between. From the obtained result, it can be revealed that the active sites of all of 5 enzymes were highly conserved (Mcwilliam et al., 2013).
Table 2: The results and Alanine racemase enzymes obtained from BLASTp.
| Access ion | Enzyme | Organism | Identit y (%) |
| WP_10 652391
6.1 |
Bifunctional UDP-N- acetylmuramoyl- tripeptide:Dalanyl-D- alanine ligase/alanine racemase | Taibaiella chishuiensis | 100 |
|
WP_11 897340 6.1 |
Bifunctional UDP-N- acetylmuramoyl- tripeptide:Dalanyl-D- alanine ligase/alanine
racemase |
Taibaiella koreensis | 82 |
|
WP_11 895176 4.1 |
Bifunctional UDP-N- acetylmuramoyl- tripeptide:Dalanyl-D- alanine ligase/alanine racemase | Taibaiella sp. F-4 | 82 |
| WP_11 099937
8.1 |
Bifunctional UDP-N- acetylmuramoyl- tripeptide:Dalanyl-D- alanine ligase/alanine racemase | Taibaiella soli | 66 |
| WP_08 610205
7.1 |
Bifunctional UDP-N- acetylmuramoyl- tripeptide:Dalanyl-D- alanine ligase/alanine racemase | Chitinopha gaceae bacterium IBVUCB1 | 64 |
| OJW79 663.1 | Bifunctional UDP-N- acetylmuramoyl- tripeptide:Dalanyl-D- alanine ligase/alanine
racemase |
Bacteroidet es bacterium 46-16 |
61 |
 |
Figure 2: Multiple sequences alignment of 5 bifunctional enzymes against the enzyme alanine racemase from chishuiensis. Yellow highlight box: active site for alanine racemase, green highlight: binding site for Dalanyl-D-alanine ligase and boxes: binding region for Dalanyl-D-alanine ligase. |
Prediction of the Secondary Structure
Analysis of secondary structure prediction from GOR on alanine racemase showed that there are 335 amino acid residues involving in the formation of the helix, 176 amino acids for extended strands (beta-sheet) formation and 300 amino acid residues in the formation of the coil, consisting of 42.72 %, 21.18 %, and 36.10 %, respectively (Figure 3) (Sen et al., 2005; Consortium, 2015; Hassan et al., 2020)
 |
Figure 3: Secondary structure prediction of alanine racemase using the GOR database. |
The 3D Structure Modeling
The 3D structure of the alanine racemase was modeled using the SWISS-MODEL database. Our enzyme (alanine racemase of Taibaiella chishuiensis) was similar to two different enzymes: UDPN-acetylmuramoyl- tripeptide-D-alanyl-D-alanine ligase (3zl8.1.A) and monomer of alanine racemase (4lut.1.A). So that our sequence from residue 18 up to 468 is similar to a template model of 3zl8, and from 468 up to 829 residues are very similar to the template model of 4lut (Figure 4).
At which about half of our sequence (from N-terminal) identity was 27.10% (with 3zl8.1.A model template), and another half of sequence (from C-terminal) identity was 39.33% (with 4lut.1.A model template).So when we combine these two template models, it will complete all residues of our sequence and it will reveal a reliable model for our sequence. Also, we modeled our sequence using Phyer2 (Figure 4). The generated structure, which was consists of one chain is very similar to the combined model of SWISS-MODELER.
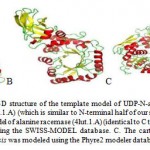 |
Figure 4: A. The cartoon 3D structure of the template model of UDP-N-acetylmuramoyl-tripeptide-D- alanyl-D-alanine ligase (3zl8.1.A) (which is similar to N-terminal half of our sequence). B. The cartoon 3D structure of the template model of alanine racemase (4lut.1.A) (identical to C terminal half of our sequence), and both were modeled using the SWISS-MODEL database. C. The cartoon 3D structure of alanine racemase from T. chishuiensis was modeled using the Phyre2 modeler database. |
Homology Model Validation
Ramachandran Plot
Ramachandran plot was used to validate the accuracy of the 3D structure by visualizing the phi (Ф) and psi (ѱ) dihedral angles of the residues of amino acids in the protein structure (Mcwilliam et al., 2013) (Figure 5).
In this case, there are 568 amino acid residues in the preferred zone, which provide 76.6 % accuracy, 127 amino acid residues (16.9 %) in the additional allowed regions, as well as 30 amino acid residues (4.0 %) in the generously allowed regions. There are 26 (3.5 %) residues of amino acid in the disallowed zones.
 |
Figure 5: Ramachandran plot of alanine racemase of T. chishuiensis predicted 3D structure. |
Verify 3D
The Verify3D software has been used to assess the consistency of the degree of every amino acid in a structure to the environment which includes the polarity. The alanine racemase predicted structure possesses an 84% of the residues with the averaged 3D-1D score >= 0.2.
From the obtained results of validation software, it is revealed that the model generated possessed a high resolution and high accuracy. Thus, the 3D model generated for alanine racemase is highly reliable (Figure 6).
 |
Figure 6: Verify 3D structure of alanine racemase of T. chishuiensis from verify 3D prediction. |
3D Structure Comparison
A comparative study was performed to investigate the structural differences of alanine racemase and the contribution to the ligand-binding cofactor on its function, based on the sequence alignment with different alanine racemase. The generated 3D structure from Phyre2 for alanine racemase of T. chishuiensis was superimposed with the structure of the templates obtained from SWISS-Model (3zl8, UDP-N-acetylmuramoyl- tripeptide-D-alanyl-D-alanine ligase, also 4lut, alanine racemase of C. difficile) (Figure7). From the superimposed structure shown in Figure 8, A it can be revealed that the catalytic residues of alanine racemase of T. chishuiensis is conserved with alanine racemase of C. difficile (PDB ID: 4lut). Also, the superimposed structure is shown in Figure 8,B revealed that the binding sites of UDP- N-acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase of Thermotoga maritima (PDB ID:3zl8) are conserved with alanine racemase of T. chishuiensis. Besides, the binding region of UDP-N acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase of T. maritima (PDB ID: 3zl8) is conserved with alanine racemase of T. chishuiensis (Figure 9).The enzyme UDP-N-acetylmuramoyltripeptide- D-alanyl-D-alanine ligase function is: ATP + UDP-N-acetylmuramoyl-L-alanyl-gamma-D- glutamyl-L-lysine + D-alanyl-D-alanine = ADP + phosphate + UDP-N-acetylmuramoyl-L-alanyl- gamma-D-glutamyl-L-lysyl-D-alanyl-D-alanine (KEGG-Enzyme) (Bhardwaj et al., 2018).
These observations show that the enzyme alanine racemase of T. chishuiensis is a bifunctional enzyme. This enzyme works with racemization of L-alanine and D-alanine as well as ligation of amino acid (Favini-stabile et al., 2013). Therefore, the enzyme alanine racemase of T. chishuiensis functionally is the same with alanine racemase of C. difficile and also has the same function with UDP-Nacetylmuramoyl-tripeptide- D-alanyl-D-alanine ligase of T. maritima. So, this enzyme has two functions that we can call it a bifunctional enzyme.
Structurally, there are differences between alanine racemase of T. chishuiensis and alanine racemase of C. difficile as well as some difference between alanine racemase and UDP- Nacetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase of T. maritima. These two parts of our enzyme are connected at the loop region: I465, H466, K467, and T468.
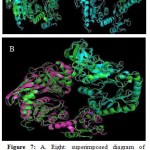 |
Figure 7: A. Right: superimposed diagram of reference UDP-N-acetylmuramoyl-tripeptide-D- alanyl-D-alanine ligase (3zl8) (green) with Phyre2 model (blue) and left: reference enzyme of Alanine racemase (4lut) (green) with Phyre2 model (blue). B. Superimposed of Phyre2 model of alanine racemase of chishuiensis (green) with reference enzyme UDPN- acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase (3zl8, pink) plus reference enzyme of monomer Alanine racemase (4lut, blue).
|
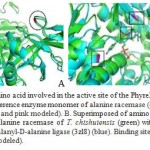 |
Figure 8: A. Superimposed amino acid involved in the active site of the Phyre2 model of alanine racemase of chishuiensis (green) with reference enzyme monomer of alanine racemase (4lut) (blue). Active sites show in the boxes (red for reference and pink modeled). B. Superimposed of amino acids involved in the binding site of the Phyre2 model of alanine racemase of T. chishuiensis (green) with reference enzyme UDP-N- acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase (3zl8) (blue). Binding sites are shown in the boxes (red for reference and pink is for modeled). |
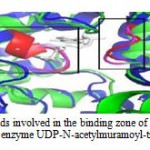 |
Figure 9: Superimposed of amino acids involved in the binding zone of the Phyre2 model of alanine racemase of chishuiensis (green) with reference enzyme UDP-N-acetylmuramoyl-tripeptide-D-alanyl |
 |
Figure 10: Sequence alignment of alanine racemase of T. chishuiensis (down sequence) with reference enzyme monomer of alanine racemase of C. difficile (4lut) (up sequence). Active sites are shown in the boxes. |
The superimposed structure of alanine racemase with binding site labeled with the reference structure (UDP-N-acetylmuramoyl- tripeptide-D-alanyl-D-alanine ligase (3zl8)) shows that the binding site was found at the similar region, overlapping each other (Figure 8,B and Figure 11). This specifies that the binding site for both alanine racemase and UDP-N- acetylmuramoyltripeptide-D-alanyl-D-alanine ligase (3zl8) was conserved and possessed similar functions.
Also, there are two binding regions. The first binding region shows less conservation and it is because of some changes of amino acids such as, the change of amino acid S to amino acid N, but both of them are small and polar so functionally both of them are the same, and also the change of amino acid T to amino acid I, which have the same characteristics, they are hydrophobic. The second binding region is strongly conserved.
 |
Figure 11: Sequence alignment of alanine racemase of T. chishuiensis (top sequence) with reference enzyme UDP- Nacetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase of T. maritima (3zl8) (down sequence). Binding sites are shown in the highlighted boxes. Also, the boxes without highlight show the binding region. |
Table 3: The active sites of the model enzyme and reference enzymes.
| The active sites of Alanine racemase, T. chishuiensis and alanine racemase, C. difficile | The active sites of Alanine racemase, T. chishuiensis plus UDP-N-acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase,
T. maritime |
| Alanine racemase, T. chishuiensis Lys | Alanine racemase, T. chishuiens is Asn 295, UDP-N- |
| 499 | acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase, T. |
| Alanine racemase, C. difficile Lys 39 | maritima Asn 264 |
| Alanine racemase, T. chishuiensis Tyr | Alanine racemase, T. chishuiensis Arg 326, UDP-N- |
| 499 | acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase, T. |
| Alanine racemase, C. difficile Tyr 725 | maritima Arg 293 |
| Alanine racemase, T. chishuiensis Lys 447, UDP-N- | |
| acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase, T. | |
| maritima Lys 405 |
The major different spots between alanine racemase of T. chishuiensis and alanine racemase of C. difficile were highlighted by using PYMOL (Figure 12, Table 3 and Table 4) and shown in red color.
 |
Table 4: The Sarface of modeled enzyme alanine racemase of T. chishuiensis and reference enzymes of alanine racemase C. difficile as well as T.maritima |
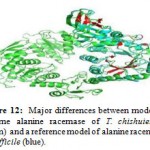 |
Figure 12: Major differences between modeled enzyme alanine racemase of T. chishuiensis (green) and a reference model of alanine racemase difficile (blue). |
The main different spots between alanine racemase of T. chishuiensis and reference enzyme UDP-Nacetylmuramoyl-tripeptide-D-alanyl-D- alanine ligase of T. maritima (3zl8) are highlighted by using PYMOL (Figure 13 and Table 4). Major different spots between two sequences are highlighted in red color.
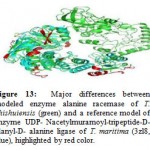 |
Figure 13: Major differences between modeled enzyme alanine racemase of T. chishuiensis (green) and a reference model of enzyme UDP- Nacetylmuramoyl-tripeptide-D-alanyl-D- alanine ligase of T. maritima (3zl8, blue), highlighted by red color. |
The predicted model of alanine racemase of T. chishuiensis from this study could serve as a useful model for further study of the structure and function of other bifunctional enzymes. Based on this study, the name of alanine racemase of T. chishuiensis can be bifunctional UDP-N-acetylmuramoyl- tripeptide: D-alanyl-D-alanine ligase/alanine racemase in the UniProt database. Besides, the structural information about alanine racemase of T. chishuiensis especially the active sites and binding sites could help drug designers to do further research. However, many scientists have been studied for the design of more specific alanine racemase inhibitors by targeting the active sites and binding sites and mutations in the enzyme strain in various microorganisms (Anthony et al., 2011; Azam and Jayaram, 2016). Due to the lack of specificity of most of the previous inhibitors, further study is required for highly selective alanine racemase inhibitors (Azam and Jayaram, 2016). Since the current study is the first study of 3D structural characterization of bifunctional alanine racemase in T. chishuiensis, it may provide useful information about the structure and function of this enzyme for drug designers to do more investigation for more specific inhibitors of alanine racemases and provide effective treatment.
Conclusion
In the current study, the bioinformatics approaches have been used to predict the 3D structure of alanine racemase (EC: 5.1.1.1) from chishuiensis. Due to the absence of a 3D structure of the bifunctional enzyme (UDP-N acetylmuramoyl-tripeptide: D-alanyl-D- alanine ligase/alanine racemase) in PDB database, we have done the homology modeling with template models of SWISS- MODELLER database (UDP-N- acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase (3zl8) plus alanine racemase (4lut)). Our enzyme (alanine racemase of Taibaiella chishuiensis) was similar to two different enzymes: UDP-N-acetylmuramoyl-tripeptide- D-alanyl-D-alanine ligase (PDB ID: 3zl8, EC: 6.3.2.10) and of alanine racemase (PDB ID: 4lut, EC: 5.1.1.1). From the sequence and 3D structure analysis, we realized that the first half of the sequence of our modeled enzyme, from residue 18 up to 468 was 27.10% identical with the existing template 3zl8, and the identity of the second half of our query from 468 up to 829 residues was 39.33%, with the template 4lut. Hence, the combination of these two template models has made a reliable model for our sequence. Both, alanine racemase from Taibaiella chishuiensis and alanine racemase of C. difficile (4lut) have the same active site at Lys (499), Lys (39), Tyr (499), Tyr (725) (Tan et al., 2014). On the other hand, both alanine racemase from Taibaiella chishuiensis and UDP-N-acetylmuramoyl-tripeptide-D-alanyl- D-alanine ligase (3zl8) from T. maritima also have the same binding site at Asn (295), Asn (264), Arg (326), Arg (293), Lys (447), Lys (405) (Dong et al., 2018). Besides, verify 3D shows good quality with a percentage of 80. Based on these observations, our enzyme (alanine racemase from T. chishuiensis) is bifunctional. Therefore, it has two commission numbers (EC: 5.1.1.1 and EC: 6.3.2.10). Findings from this study may help to contribute to the rational design of the remaining unknown structure of the same enzyme in other organisms and assist in designing proteins model with enhanced properties.
Acknowledgments
I wish to express my sincere appreciation to my main lecturer, Prof. Dr. Mohd Shahir Shamsir Omar, for encouragement, guidance, critics, and friendship. Without his continued support and interest, this study would not have been the same as presented here. I am also indebted to the Ministry of Higher Education Afghanistan and Higher Education Development Program (HEDP), for supporting and funding this study. Special thanks to the University Technology Malaysia (UTM) for providing the opportunity to study at this prestigious university.
References
- Muhammad and B. Zhao, “Purification , Characterization and Inhibition of Alanine Racemase from a Pathogenic Strain of Streptococcus iniae,” Polish J. Microbiol., vol. 68, no. 3, pp. 331–341, 2019.
- Muhammad, Y. Li, S. Gong, Y. Shi, and J. Ju, “Cloning , Biochemical Characterization and Inhibition of Alanine racemase from Streptococcus iniae,” bioRxiv, no. 611251, pp. 1– 25, 2019.
- A. Wood and C. . Gunsalus, “d-Alanine Formation: A Racemase In Streptococcus Faecalis,” ournal Biol. Chem., vol. 190, no. 1, pp. 403–416, 1959.
- S.Liu et al., “Function of alanine racemase in the physiological activity and cariogenicity of Streptococcus mutans,” Sci. Rep., vol. 8, no. 30, pp. 1–8, 2018.
- M. S. Islam, A. Aryasomayajula, and P. R. Selvaganapathy, “A Review on Macroscale and Microscale Cell Lysis Methods,” Micromachines, vol. 8, no. 83, pp. 1–27, 2017.
- T. Nomura, I. Yamamoto, F. Morishita, Y. Furukawa, and O. Matsushima, “Purification and Some Properties of Alanine Racemase From a Bivalve Mollusc Corbicula japonica,” J. Exp. Zool., vol. 9, no. 289, pp. 1–9, 2001.
- I .Schomburg et al., “BRENDA , the enzyme database : updates and major new developments,” Nucleic Acids Res., vol. 32, no. 4, pp. 431–433, 2004.
- Y .Wei et al., “Alanine racemase is essential for the growth and interspecies competitiveness of Streptococcus mutans,” Int. J. Oral Sci., vol. 8, no. 4, pp. 231–238, 2016.
- H .Im, M. L. Sharpe, U. Strych, M. Davlieva, and L. Krause, “The crystal structure of alanine racemase from Streptococcus pneumoniae, a target for structure-based drug design,” BMC Microbiol., vol. 11, pp. 1–15, 2011.
- K .G. Anthony et al., “New classes of alanine racemase inhibitors identified by high- throughput screening show antimicrobial activity against mycobacterium tuberculosis,” PLoS One, vol. 6, no. 5, pp. 1–9, 2011.
- M. A. Azam and U. Jayaram, “Inhibitors of alanine racemase enzyme: A review,” J. Enzyme Inhib. Med. Chem., vol. 31, no. 4, pp. 517–526, 2016.
- X. Tan, R. Zhang, T. Meng, H. Liang, and J. Lv, “Taibaiella chishuiensis sp . nov ., isolated from freshwater,” Int. J. Syst. Evol. Microbiol., vol. 64, pp. 1795–1801, 2014.
- R. M. Couñago, M. Davlieva, U. Strych, R. E. Hill, and K. L. Krause, “from Bacillus anthracis ( Ames ),” BMC Struct. Biol., vol. 15, pp. 1–15, 2009.
- H. Dong et al., “Biochemical and Biophysical Research Communications Enzymatic characterization and crystal structure of biosynthetic alanine racemase from Pseudomonas aeruginosa PAO1,” Biochem. Biophys. Res. Commun., vol. 503, no. 4, pp. 2319–2325, 2018.
- J. Ju et al., “Correlation between catalytic activity and monomer — dimer equilibrium of bacterial alanine racemases,” J. Biochem., vol. 149, no. 1, pp. 83–89, 2011.
- O. A. Asojo, D. A. Hyman, and M. Monica, “Structural and biochemical analyses of alanine racemase from the multidrug-resistant Clostridium difficile strain 630,” Biol. Crystallogr., vol. 70, no. 7, pp. 1922–1933, 2014.
- P. Chen et al., “Structure of the full-length Clostridium difficile toxin B,” Nat. Struct. Mol. Biol., vol. 26, no. 8, pp. 712–719, 2019.
- S. Favini-stabile, C. Contreras-martel, N. Thielens, and A. Dessen, “MreB and MurG as scaffolds for the cytoplasmic steps of peptidoglycan biosynthesis,” Environ. Microbiol., vol. 15, no. 12, pp. 3218–3228, 2013.
- E. Gasteiger et al., “ExPASy : the proteomics server for in-depth protein knowledge and analysis,” Nucleic Acids Res., vol. 31, no. 13, pp. 3784–3788, 2003.
- S. Hassan, J. Javid, A. Sadiqua, N. Fatima, A. Mohsin, and H. Akram, “Thyroid Cancer TC-1 : an insight from 3D structure prediction to virtual screening for computational drug design,” Biomed. Lett., vol. 6, no. 1, pp. 127–137, 2020.
- T. Z. Sen, R. L. Jernigan, J. Garnier, and A. Kloczkowski, “Structural bioinformatics GOR V server for protein secondary structure prediction,” Bioinformatics, vol. 21, no. 11, pp. 2787–2788, 2005.
- M. Biasini et al., “SWISS-MODEL : modelling protein tertiary and quaternary structure using evolutionary information,” Nucleic Acids Res., vol. 42, no. W1, pp. W252–W258, 2014.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass, and M. J. E. Sternberg, “The Phyre2 web portal for protein modeling , prediction and analysis,” Nat. Protoc., vol. 10, no. 6, pp. 845– 858, 2015.
- P. D. O. De Meo and D. Cozzetto, “The PMDB Protein Model Database,” Nucleic Acids Res., vol. 34, no. suppl_1, pp. 306–309, 2006.
- G. Janson, C. Zhang, M. G. Prado, and A. Paiardini, “Structural bioinformatics PyMod 2 . 0 : improvements in protein sequence-structure analysis and homology modeling within PyMOL,” Bioinformatics, vol. 33, no. 3, pp. 444– 446, 2017.
- U. Consortium, “UniProt : a hub for protein information,” Nucleic Acids Res., vol. 43, no. D1, pp. D204–D212, 2015.
- H. Mcwilliam et al., “Analysis Tool Web Services from the EMBL-EBI,” Nucleic Acids Res., vol. 41, no. W1, pp. 597–600, 2013.
- M. Kouza and E. Faraggi, “Chapter 2,” Humana Press. New York, NY., vol. 1484, pp. 7–24, 2017.
- M. Wiltgen and G. G. Hospital, Algorithms for Structure Comparison and Analysis : Homology Modelling of Proteins, vol. 1. Elsevier Ltd., 2018.
- T. Bhardwaj and P. Somvanshi, “Gene Reports A computational approach using mathematical modeling to assess the peptidoglycan biosynthesis of Clostridium botulinum ATCC 3502 for potential drug targets,” Gene Reports, vol. 12, pp. 179–186, 2018.

This work is licensed under a Creative Commons Attribution 4.0 International License.



