

How to Cite | Publication History | PlumX Article Matrix
Pritee Chunarkar Patil*  ,Darshit Ashokkumar Patel, and Vidya Sunil Tale
,Darshit Ashokkumar Patel, and Vidya Sunil Tale
Department of Bioinformatics, Rajiv Gandhi Institute of IT and Biotechnology, Bharati Vidyapeeth (Deemed to be University), Pune, Maharashtra, India -411046.
Corresponding Author E-Mail: preeti.chunarkar@bharatividyapeeth.edu
DOI : http://dx.doi.org/10.13005/bbra/2889

ABSTRACT:
Advancement in sequencing techniques and transformative progress in metagenomics provides an unprecedented platform for functional and taxonomic characterization of the enormous microbial diversity inhabiting and governing various biochemical processes of the freshwater sources. Metagenomic analysis of freshwater resources has led to the discovery and identification of novel microbial genes and an understanding of how microorganisms mediate energy and carbon. In this study, we report the taxonomical classification of bacterial sequences obtained from 6 dam reservoir sites in Pune city, Maharashtra, India. The analysis was performed using two different alignment tools: BLAST and Kaiju. The bacterial diversity was dominated by the presence of Vogecella indigofera, uncultured Proteobacterium, Wolinella Succinogenes, Chromobacterium violaceum, and Heliobacter billis. It was further observed that, despite an identical bacterial composition over various reservoir sites, there were nominal differences in the relative abundance of the inhabitant species. Almost all reservoirs were dominated by Vogecella indigofera (~29%) and uncultured Proteobacterium (~15%). A seasonal analysis performed using BLAST resulted in a number of species exclusive to the season and the site of their growth. A high proportion of unidentified sequences were also reported which demands sequential identification. The results obtained through BLAST and Kaiju, were significantly different, suggesting inconsistencies and inaccuracies in existing metagenomic reads comparison.
KEYWORDS: Bacterial Community; BLAST; Environmental Microbiology; Fresh water Metagenomics; Kaiju Tool; Metagenomics
Download this article as:| Copy the following to cite this article: Patil P. C, Patel D. A, Tale S. V. Metagenomic Analysis of Dam Reservoirs in Pune City for Bacterial Fingerprints Through BLAST and Kaiju Tool. Biosci Biotech Res Asia 2021;17(4). |
| Copy the following to cite this URL: Patil P. C, Patel D. A, Tale S. V. Metagenomic Analysis of Dam Reservoirs in Pune City for Bacterial Fingerprints Through BLAST and Kaiju Tool. Biosci Biotech Res Asia 2021;17(4). Available from: https://bit.ly/3oZXSc0 |
Introduction
Until the late 20th century, the outcomes of microbiology were restricted by the meagre potential of pure culturing techniques in terms of throughput data and accuracy. This is largely because of the dependency of culturing techniques on the ability of a microbe to grow in the appropriate medium. Although culturing techniques have led to the discovery of a plethora of microorganisms, a century worth of research has only able to identify 1% of the speculated microbial population. Acknowledging the limitations of pure culturing techniques, Pace and colleagues utilized the available sequencing techniques to sequence the 16s rRNA and 5s rRNA in order to describe the microbial diversity in an environmental soil sample 1. This was one of the pioneer projects that led to the development of a new field of environmental ecology, called metagenomics. First introduced by Jo Handlesman, metagenomics is an innovative approach of analyzing the total genetic material from an environmental source in order to understand the taxonomic composition and the role of each member in the community 2. Simultaneously, advancement in sequencing technologies leading to Next Generation Sequencing (NGS), had a remarkable effect on the frequency and accuracy of the studies performed under the domain of metagenomics. Since the start of the 21st century, efforts towards metagenomic analysis have increased several folds with multiple benchmark projects aimed at understanding the complex taxonomic structure of the microbial communities inhabiting various environmental sources and their role in maintaining the biogeochemical cycles responsible for governing the flow of carbon and energy in the ecosystem. Some of the most remarkable studies have been published covering a wide range of environmental samples that are directly or indirectly involved in the health of our species. One of the most interesting of these studies started in 2003 was “The Sorcerer II Global Ocean sample expedition” (popularly known as GOS) that followed the renowned route of Darwin’s HMS Beagle, answering one of the most complex questions regarding the diversity of microbes in global marine environments (data sets of which are available on NCBI and CAMERA databases) 3. The uncultured viral community associated with the human fecal matter was another vital study carried out by 4 under the paradigms of metagenomics revealing important data regarding ~60 strains of phages and other viruses colonizing the lower areas of the gut microbiome. Another set of studies deeply investigates local environmental samples, examples of which include acid mine drainage biofilms, Sargasso Sea, Whale falls, Eel river sediments, Minnesota farm soil, Pleistocene bear caves fossils, Soudan Mine, Hawaii Ocean, Mammoth fossils, Bras del Port Saltern, Coral holobiont, Neanderthal microbiome, Mediterranean Sea, Coral Reefs in Australia, Tasmanian Tiger Genome and many more. The collection of data from these studies has enhanced our understanding of various factors of human evolution, gut microbiome, and most importantly the role of microorganisms in the environmental samples that are directly involved in the general human lifestyle of that particular locality 5. The data collected from the various samples can be categorized as either sequence driven data or function-driven data. While the former has a great potential to discover novel microbial species and their evolutionary relationship with existing taxa, the latter reveals important features about the genetic and proteomic factors responsible for carrying out the physiochemical processes within a particular microbial community. Considering the wide success of various large-scale environmental projects, Metagenomics has inevitably permeated in the research of freshwater bodies and related sciences like limnology to address the lacunae in our understanding of the microbial factors responsible for 90% of global organic degradation and nutrient recycling 6.
Different kingdoms of microorganisms are known to inhabit all kinds of extreme and non-extreme environments. Similarly, each freshwater body has its indigenous microbiome or microbial community that might not occur in other systems like marine, soil, or terrestrial ecosystems 7. Microorganisms from all kingdoms like protozoa, fungi, bacteria, and archaea present in these freshwater ecosystems dominate a large number of biogeochemical processes that occur in the respective habitat. Most of the members of this community are also directly involved in maintaining the quality of water and pollutants. Bacterial species (Gram-positive and negative) are one of the most frequent inhabitants of freshwater bodies (more than 90%) like rivers, lakes, and other inland waters 8. Studies by 9 further confirmed that bacterial species found in a particular freshwater system is closely related to communities from other freshwater sources but differed significantly when compared to marine ecosystems. They also established that certain bacterial colonies are typical to the freshwater systems and were universally observed in different freshwater systems, examples of such families of bacteria include Actinobacteria (ACK M1), Betaproteobacteria (GKS98, polynucleobacter, and R-BT065), and Bacteroides (SOL). It is also observed in many species that despite the cosmopolitan presence of certain families of bacteria in different climatic zones, they did show adaptation to a range of temperature but could only acclimatize to the particular temperature they are subjected to 10. Furthemore, freshwater habitats also provide the essential conditions to favor the growth of a range of bacterial pathogens like Botulism, Campylobacteriosis, Cholera, E. coli Infection, M. marinum infection, Dysentery, Legionellosis (two distinct forms: Legionnaires’ disease and Pontiac fever), Leptospirosis, Otitis Externa (swimmer’s ear), Salmonellosis, Typhoid fever and Vibrio Illness 11. These pathogens are directly responsible for an estimated 1.8 million death (4.1% of the global burden of diseases) globally. Consequently, the uses of antibiotics by a large population contribute to the development of antibiotic-resistant genes in the bacterial colonies through fecal matter and industrial contamination.
Advancement in sequencing techniques and transformative progress in metagenomics provides an unprecedented platform for functional and taxonomic characterization of the enormous microbial diversity inhabiting and governing various biochemical processes of the freshwater sources. Metagenomic analysis of freshwater resources has led to the discovery and identification of novel microbial genes and an understanding of how microorganisms mediate energy and carbon. A combination of techniques developed under the paradigms of functional metagenomics and sequence-driven metagenomics has been utilized in identifying novel microbial species and their peculiar biogeochemical processes, enzymatic or catalytic properties, or their pathogenic properties that are primary markers in deciding the quality of water. Various projects recently carried out in the concerned field have led to the discovery of antibiotic-resistant bacterial strains, novel viral families, gene families, and pathogenic strains. The freshwater metagenomics approach further provides the potential to compare bacterioplankton of different water bodies, identifying generic microbial families of specific water body systems (lake, river, pond) and phylogenetic classification of resident microbial members. Most importantly, Metagenomics has extensively helped in categorizing genes from various bacterial strains that are directly involved in antibiotic resistance under a new term, called resistome 12.
Realizing the potential and success of metagenomics in decoding microbial communities from local freshwater resources, we focused this study on the analysis of 65 samples collected over 6 dams (Khadakwasla, Panshet, Varasgaon, Bhatgar, Mulshi, and Temghar) in Pune city (Maharashtra, India) to elucidate the bacterial composition and establish their taxonomic relationship. The building of river dams has a significant effect on the riverine environment causing a myriad of physical, biological, and chemical variations. These effects are key factors in deciding the composition of the microbial communities inhabiting within them as discussed before 13. We further analyzed, samples from the same sources over various seasons (Summer, Winter, and Monsoon), and from the surface, middle and, bottom layers. The seasonal analysis was carried out with an objective to understand the effect of the annual hydrological cycle on the bacterial community while the water column was analyzed to draw important conclusions regarding the effect of light and temperature on the bacterial composition.
Material and Methods
Chemicals
All the bulk chemicals, solvents used in the study were of analytical reagent (AR) grade and procured from suppliers including SD fine-chemicals, Loba Chemie, Sisco Research Laboratories and Qualigenes, India.
Fine chemicals like sodium acetate, STE buffer, lysozyme, proteinase K, dithiothrietol, HEXfluorescent dye were obtained from Sigma–Aldrich, Germany, and geneOmbio Technologies, India. Membrane filters of 45µm were procured from Himedia, India. DNA ladder mix, RNAase was procured from Promega, UK; dNTP, HF PCR buffer, Taq polymerase, was PCR primers were provided by IDT, USA and PCR purification kit was purchased from geneOmbio Technologies, India.
Water sample Collection
Total 72 water samples were collected from six dams, Temghar, Bhatgar, Khadakwasla, Warasgaon, Panshet, and Mulshi of Pune city (18.5204° N, 73.8567° E). This study had been taken three samples (Bottom layer, Middle layer, and Upper layer) at a time for every season (rainy, winter, and summer). Total of nine samples were taken from one place. All water samples were collected in a sterile Labifie borosilicate glass reagent bottle with a screw cap. While taking the sample, the bottle was used to rinse three times with the respective sample (river/lake/dam water). After rinsing, the bottle was submerged below the water level till it will fill with the sample below 2.5 cm below the lid to maintain homogeneity. After the collection of the samples, till analysis, it is used to store at 400C in the fridge.
DNA isolation from water samples
15ml samples were collected and stored in sterilized bottles followed by the addition of 1.5ml 3M Sodium acetate and 33ml absolute ethanol. The mixture was stored at 40C until extraction.
For DNA extraction, the mixture was centrifuged at 10000 rpm for 20 mins (60C) and the supernatant was discarded. The pellet was washed and re-suspended in 0.9% saline. Nucleic acid extraction was performed by the addition of the STE buffer, lysozymes, and Proteinase K (10µg/ml). Purification of nucleic acid was performed by the NaCl and SDS lysis followed by phenol: chloroform: isoamyl alcohol-based extraction. Extracted and purified DNA was deproteinized thrice using the Tris-saturated phenol (isoamyl alcohol: CHCl3: Phenol in ratio 2:48:50), followed by CHCl3: isoamyl alcohol (24:1). The DNA was precipitated using 2% sodium acetate and absolute alcohol. The DNA was dried and dissolved in 30L nuclease-free water.
DNA Sequencing
The extracted DNA was amplified using PCR and the amplicons were used for sequencing. The sequencing of DNA was performed using the BigDye Terminator V 3.1 Cycle sequencing kit (Applied Biosystems, USA); 2µl Sequencing primer (1.6pMol), 2µl Purified PCR product (template DNA ~ 50 -100 ng/uL concentration), 1.8µl sequencing buffer, 0.5µl ready reaction mix and 3.7µl Nuclease free water.
Applied Biosystems 3130 Genetic Analysis (Automated Sequencing Analysis machine) was used for the analysis of the sequencing products obtained from the cycling kit. The sequences were generated in .abl format which was then exported to FASTA format for analysis on BLAST and Kaiju tools.
Results and Discussion
72 samples collected from 6 different dam sites were compared and analyzed to the existing databases using BLAST and Kaiju tools for analysis of bacterial composition. It was observed that sequences from Burkholderia, Sutterella, Wolinella, Escherichia, Acetobacter, and Staphylococcus were relatively abundant compared to the others, while an average of 40% nucleotide sequences obtained from the various samples remained unidentified following the comparison with the mentioned database. It is a clear indication of uncultured bacterial communities. A consistent bacterial composition was observed over various dam reservoirs at different locations in the city.
Analysis Through Kaiju Tool
Figure 1 to 6, shows the relative abundance of different bacterial groups within each of the reservoir systems. Among the 6 dam reservoir systems, Khadakwasla Dam has the highest proportion of unidentified sequences at 49% while Varasgaon Dam has the lowest percentage of unidentified sequences at 35%. While all the systems resulted in a relatively higher majority of Burkholderia (8-11%), Sutterella (8-14%), Wolinella (5-9%), Staphylococcus (3-7%), Acetobacter (4-7%), Lactobacillus (2-4%), and Escherichia (5-8%), a few cases were observed to have a significantly lower population of the mentioned species, for example, Mulshi dam comprising 7% Burkholderia, Panshet comprising 3% Wolinella and Khadakwasla comprising only 2% Escherichia sequences. Albeit, in lower concentrations, another set of sequences observed throughout the 6 systems were from Pleomorphonas (1-4%), Helicobacter (0-3%), Rhodococcus (0-2%), Coxiella (1-2%), Taylorella (1-3%), Vibrio (0-2%), and Thiomonas (2-3%). With an exception of Mulshi Dam, Campylobacter was observed in all the other systems at relatively low concentrations ranging from 0-1%. Paenobacillus was completely absent in Khadakwasla and Varasgaon dam while ranging at 0-1% in other systems. Similar trends were observed for other sequences from Paenathrobacter (0-1%), Salmonella (0-1%), Curvibacter (0-2%), and Bifidobacterium (0-1%), where each of these species was absent from 2 reservoir systems: Paenathrobacter from Temghar and Varasgaon dam, Salmonella from Panshet and Varasgaon Dam, Curvibacter from Panshet and Varasgaon Dam, and Bifidobacterium from Bhatgar and Temghar Dam. Sequences from the Bacillus group were only obtained from Bhatgar and Khadakwasla Dam in ultra-trace quantities ranging from 0-1%. Mycobacterium sequences were further observed to be absent from Khadakwasla, Temghar, and Panshet dam, while Vitrioscella sequences were missing from Bhatgar, Mulshi, and Temghar Dams. Furthermore, Varasgaon Dam and Mulshi Dam reported the presence of sequences from exclusive groups, Listeria and Klebsiella, respectively.
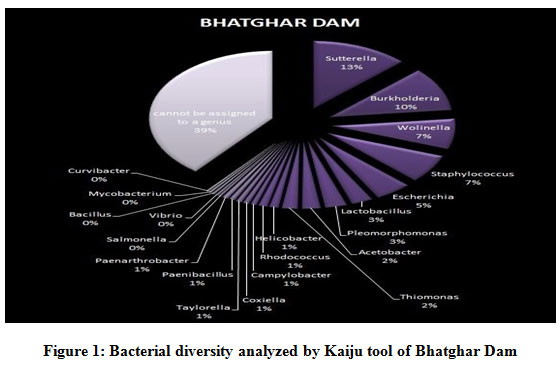 |
Figure 1: Bacterial diversity analyzed by Kaiju tool of Bhatghar Dam. |
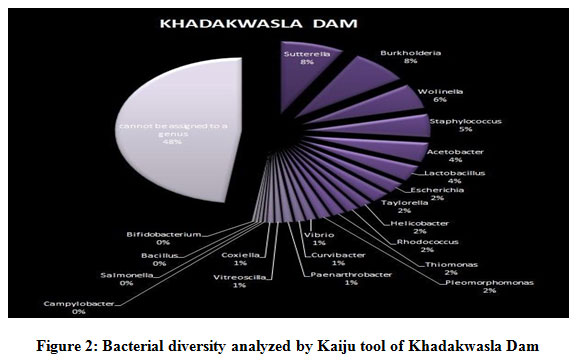 |
Figure 2: Bacterial diversity analyzed by Kaiju tool of Khadakwasla Dam. |
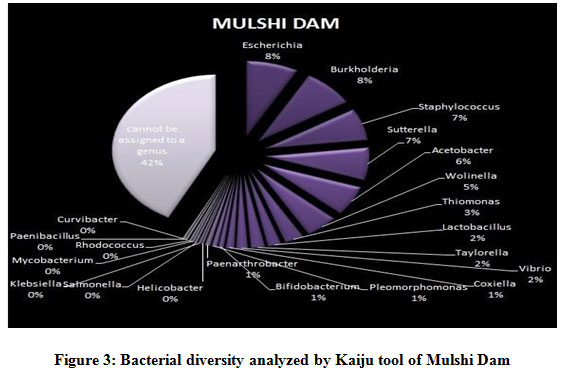 |
Figure 3: Bacterial diversity analyzed by Kaiju tool of Mulshi Dam. |
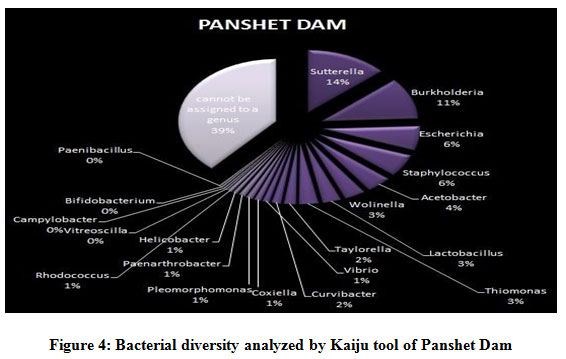 |
Figure 4: Bacterial diversity analyzed by Kaiju tool of Panshet Dam. |
The presence of the Staphylococcus aureus and Burkholderiales cepacia complexes, suggests an important link to the human respiratory tract as the former is a commensal in the microbiome while the latter is an opportunistic pathogen causing respiratory infections 14, 15. Burkholderia genus comprises a range of 30 different species that are involved in various bioremediation, ecological, and pathogenic processes. Some of the species from Burkholderia have been known pathogens of plants (carnation, onions, blight) and have also found early applications as bioweapons (Germany, during World War I) for its ability to cause infectious diseases in humans, horses, mules, and donkeys like melioidosis, septicemia, and pneumonia 16, 17. Furthermore, Burkholderia species have been actively utilized in industrial bioremediation processes, nutrition delivery in plants, and disintegration of biohazardous waste 18. A high percentage presence (~18%) of Suterella sp. also indicates fecal contamination since the bacterial species has been most commonly isolated from human feces 19. Similarly, a considerable composition of reservoir freshwater bacterial colonies was identified to be commensals of the human GI tract microbiome like various strains of Escherichia coli, while sequences of various pathogenic strains from the family Campylobacter like Wolinella succinogenes and Helicobacter cinaedi were recovered in trace quantities 20, 21, 22. Escherichia also shows a peculiar ability to grow under low nutrient and other physiological stress, therefore, showing a high species richness. It also contributes to the microbial resistome of the habitat by expressing various genes for multidrug antibiotic resistance 23. Various pathogenic species of the Helicobacter genus including H. bilis, H. canis, H. trogontum, and H. hepaticus responsible for causing a range of infections in mammals were also persistently recovered throughout the year, although in trace quantities 24, 25, 26. Coxiella burnteii, a Rickesitta looking bacteria, further adds to the list of pathogens recovered from the river samples. It is a characteristic mild pathogen, resistant to extreme environmental changes and is the sole etiological factor responsible for the Q fever in cattle and humans 27.
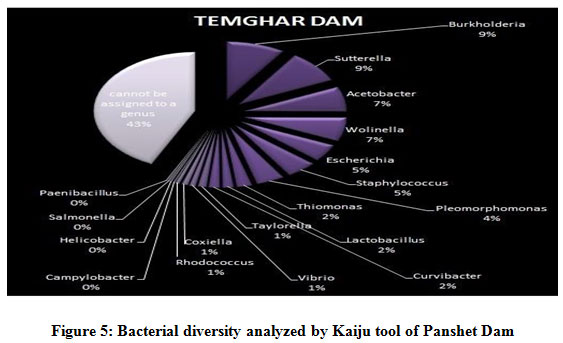 |
Figure 5: Bacterial diversity analyzed by Kaiju tool of Panshet Dam |
 |
Figure 6: Bacterial diversity analyzed by Kaiju tool of Varasgaon Dam |
Analysis Through BLAST
A seasonal analysis was performed for each reservoir site was performed using BLAST to understand the effect of the annual hydrological cycle on the bacterial composition. BLAST analysis resulted in a much more sensitive taxonomic analysis compared to that of Kaiju with significant differences in the bacterial composition. The results obtained through BLAST alignment reported various species from the bacterial groups identified in the Kaiju analysis, for example, Wolinella succinogenes, Helicobacter billis, Helicobacter pylori, and Bacillus pumilus among others. Additionally, sequences from bacterial taxa, Vogisella, Chromobacterium, Psuedogulbekiania, Citrobacter, Flexispira, Streptomyces, Neisseria, Pantoea, Enterobacterium, Glyxomyces, and Iodobacter were also identified from various samples suggesting the presence of a wide spectrum of ecologically important bacteria in the reservoir sites. Figure 7-12, shows the relative abundance of various species obtained from different sites in Summer, Winter, and Rainy seasons.
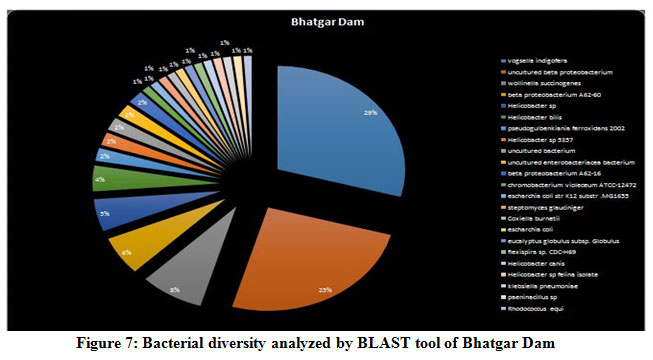 |
Figure 7: Bacterial diversity analyzed by BLAST tool of Bhatgar Dam |
 |
Figure 8: Bacterial diversity analyzed by BLAST tool of Khadakwasala Dam |
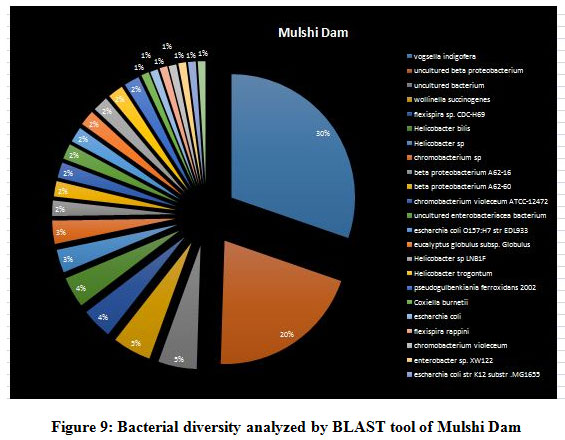 |
Figure 9: Bacterial diversity analyzed by BLAST tool of Mulshi Dam |
According to the BLAST analysis, Vogisella indigofera was the most dominating bacterial species found among all the dams. Vogisella indigofera is a known mesophilic freshwater proteobacterium that shows a perennial presence in relatively high concentrations in rivers 28. In comparison to other seasons, winter can be attributed to a rise in the population of Wolinella Succiongenes in both the rivers that might possibly affect the anaerobic consumption of sulfites, polysulfide, nitrites, fumarates, and nitrates 29. One of the rare bacteria, Chromobacterium violaceum, was slightly more abundant during the rainy season compared to the summer or winter. C. violaceum is common to soil or stagnant waters, therefore, its presence can be directly correlated with the interplay of rivers with the environmental soil, ponds, or lakes. While ingestion of water contaminated by C. violaceum might not be harmful, it can cause localized skin infections with a potential to progress into septicemia, hence increasing the chances for multiorgan failure 30. A high percentage of bacterial sequences obtained from unknown species of Proteobacteria phylum or Enterobacteriaceae family might have a potential role in maintaining ecological cycles involving nitrogen, oxygen, sulfur, and carbon. Another agent of contamination, Bacillus pumilus was reported from Bhatgar and Khadakwasla dam. Bacillus pumilus is a Gram-positive aerobic bacteria that is commonly an inhabitant of soil microbial communities and is actively involved in rhizosphere formation and fungal resistance in various plants like red peppers and wheat 31. Furthermore, it serves the ecology of the surrounding environment by taking the responsibility for fixing atmospheric nitrogen to ammonia 32. It is also rarely pathogenic to humans causing toxic effects on epithelial cells through a complex of lipopeptide and pumilacidins. Streptomyces glaucinger represents the cosmopolitan mesophilic freshwater actinomycete that actively degrades starch, xanthin, casein, and hypoxanthine. It has applications in anti-fungal therapies for its pathogenic activity towards clinically important Candida albicans 33. Industrially important strains of bacteria were also found to be active members of the Pune dam microbiome community, like, Pantoea agglomerans, a species that can be exploited as a rich source for various antibiotics like phenazine and pantocins 19. Furthermore, P. agglomerans have been recently modified to cause antimalarial effects towards Plasmodium in mosquito guts 34.
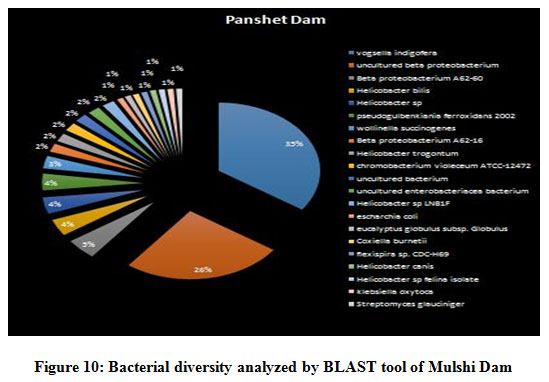 |
Figure 10: Bacterial diversity analyzed by BLAST tool of Mulshi Dam |
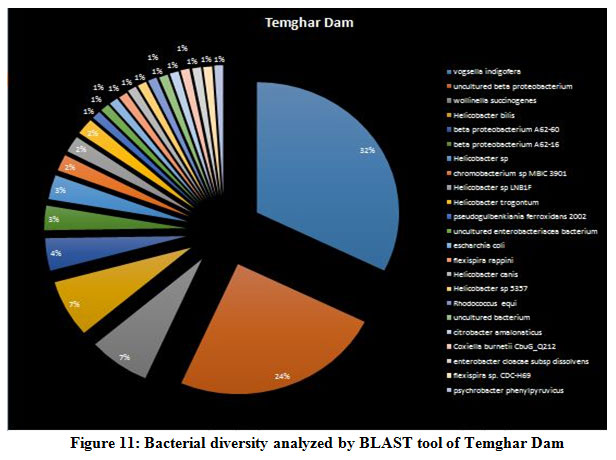 |
Figure 11: Bacterial diversity analyzed by BLAST tool of Temghar Dam |
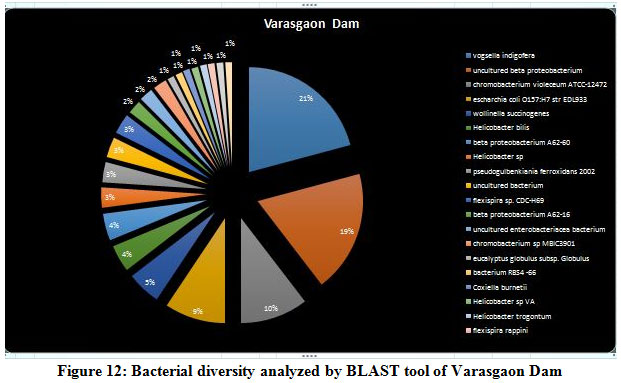 |
Figure 12: Bacterial diversity analyzed by BLAST tool of Varasgaon Dam |
There were certain bacterial species exclusive to certain dams in specific seasons. An unusual presence of Psychrobacter pacificensis was reported in Khadakwasla dam during the rainy season, as this is a non-motile, deep water bacteria adapted to psychrophilic conditions that show oxidative and catalytic activities 35. The summer season further marks the growth of Zooglea ramigera in Khadakwasla dam, suggesting deposition of minerals, since Z. ramigera growth indicates the presence of an enriched aqueous environment 36. Panshet dam accounts for on of the highest species richness among all the environments studied. In addition to species obtained in other resources, Panshet dam sustains the growth of Acidiferrobacter thiooxydans, Achromatium, and Neisseria elongata. A. thiooxydans are a highly resistant species towards acidic and thermal stress belonging to the family, Ectothiorhodospiraceae. It might have a potential role in the anaerobic oxidation of Iron and Sulfur radicals, hence contributing to the overall ecology of the environment 37. Another sulfur oxidising class of bacterial genus called, Achromatium was also isolated from Panshet Dam. Achromatium has an interesting genome that is highly similar to many environmental genomes and often contributes to the bacterial population at the interface of river sediments 38. An ancestor of the pathogenic Neisseriaceae family, N. elongata was also isolated from Panshet Dam. Usually a commensal in the pharynx of humans, these organisms can otherwise cause systematic diseases like endocarditis, osteomyelitis, and septicemia 39. A biologically and industrially important genus, Vitrioscella, was isolated from Mulshi dam during the rainy season. It is one of the earliest bacteria that has the capability to biosynthesize bacterial hemoglobin and find many applications in industrial biotechnology for its ability to promote cell growth, fermentation, biodegradation of toxic substances, and expulsion of toxic xenobiotics from cells, enhance protein synthesis, and metabolic productivity 40. A chemoorganoheterotrophic bacterium, Leptothrix fluviatis was isolated from Temghar dam, indicating neutral to slightly acidic environmental conditions. Leptothrix further suggests the presence of rust contamination in the dam as the organism is highly ferrugenic 41.
Conclusion
A deep metagenomic investigation of bacterial composition at various dam reservoir sites in Pune resulted in the identification of various etiologically, industrially, and ecologically important bacterial strains. However, an analysis performed using tools, Kaiju, and BLAST, reported significant differences in the taxonomic classification obtained. This suggests a lot of inaccuracy and inconsistency in results obtained through the existing tools for the alignment of metagenomic reads. To develop an efficient metagenomic analysis technique is the need of the hour.
Acknowledgment
We would like to acknowledge our institute for providing us the infrastructure facilities. We would like to thank Ms. Sanober Khatoon for helping us in technical issues.
Conflict of Interest
Authors declare no conflict of interests.
Funding Source
Bharati Vidyapeeth (Deemed to be University) has provided the funding for the current project.
References
- Wooley J, Godzik A, and Friedberg I. A primer on Metagenomics. PLoS Computational Biology.. 2010. 6(2) : e100067
CrossRef - Handlesman J, Rondon M, Brady S, Clardy J, and Goodman R. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chemistry and Biology. 1998. 5(10) :
CrossRef - Rusch D, Halpern A, Sutton G, Heidelberg K, Williamson S, et al. The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biology 5(3) :77.
- Breitbart M, Hewson I, Felts B, Mahaffy J, Nultan J, et al. Metagenomic Analyses of an Uncultured Viral Community from Human Feces. Journal of Bacteriology. 2003. 185 (20) :6220.
CrossRef - McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, et al.. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010. 20(9) :
CrossRef - Wetzel R. Freshwater ecology: changes, requirements, and future demands. Limnology. 1: 3.
CrossRef - Shreeve J. The blueprint of life. US News World Reports. 139(16) :70.
- Hiorns W, Methe´ B, Nierzwickibauer S, and Zehr J. Bacterial diversity in Adirondack mountain lakes as revealed by 16S rRNA gene sequences. Applied Environmental Microbiol 1997. 63: 2957.
CrossRef - Zwart G, Crump B, Kamst-van A, Hagen F, and Han S. Typical freshwater bacteria: an analysis of available 16S rRNA gene sequences from plankton of lakes and rivers. Aquat Microb Ecol. 2002. 28:141-155
CrossRef - Hahn M, and Pockl M. Ecotypes of planktonic Actinobacteria with identical 16S rRNA genes adapted to thermal niches in temperate, subtropical, and tropical freshwater habitats. Applied Environmental Microbiology. 71: 766.
CrossRef - Sharma S, Sachdeva P, and Virdi J. Emerging water-borne pathogens. Applied Microbiol Biotechnolgy. 61 : 424.
CrossRef - Nnadozie C, and Odume O. Freshwater environments as reservoirs of antibiotic resistant bacteria and their role in the dissemination of antibiotic resistance genes. Environmental pollution. 2009. 254(part B) :113067.
CrossRef - Yan Q, Bi Y, Deng Y, He Z, Wou L, et al. Impacts of the Three Gorges Dam on microbial structure and potential function. Scientific Reports. 2015. 5:
CrossRef - Masalha M, Borovok I, Schreiber R, Aharonowitz Y, and Cohen G . Analysis of transcription of the Staphylococcus aureus aerobic class Ib and anaerobic class III ribonucleotide reductase genes in response to oxygen. Journal of Bacteriology. 2001. 183 (24): 7260.
CrossRef - Chandler L. Challenges in Clinical Microbiology Testing. Accurate results in ththe clinical laboratory. 315.
CrossRef - Godoy D, Gaynor R, Simpson A, Aanensen DM, Pitt T, et al. Multilocus sequence typing and evolutionary relationships among the causative agents of melioidosis and glanders, Burkholderia pseudomallei and Burkholderia mallei. Journal of Clinical Microbiology. 2003. 41(5) :2068.
CrossRef - Vazquez N, Cesar V, Rocio A, Yendi N, Marco L, et al. Modifications of bacterial populations in anthracene contaminated soils. Applied soil ecology. 61 :113.
CrossRef - Coenye T. and Vandamme P. Diversity and significance of Burkholderia species occupying diverse ecological niches. Environmental Microbiology. 2003. 5(9) :719.
CrossRef
- Wang L, Christophersen C, Sorich M, Gerber J, Angley M, et al. Increased abundance of Sutterella spp. and Ruminococcus torques in feces of children with autism spectrum disorder. Molecular Autism. 2013. 4(1) :42.
CrossRef - Baar C, Eppinger M, Raddatz G, Simon J, Lanz C, et al. Complete genome sequence and analysis of Wolinella succinogenes. Proceedings of the National Academy of Sciences of the United States of America. 100(20): 11690.
CrossRef - Batt C. Alcaligenes. Encyclopedia of Food Microbiology Second edition. 2014 :38.
CrossRef - Kawamura Y, Tomida J, Morita Y, Fujii S, Okamoto T, et al.. Clinical and bacteriological characteristics of Helicobacter cinaedi infection. Journal of Infection and Chemotherapy. 2014. 20(9) : 517.
CrossRef - Kaper J, Nataro J and Mobley H. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004. 2 :123.
CrossRef - Dutasta F, Samaha E, Carayol N, Masse J, Bourillon C, et al.. Acute Colitis Caused by Helicobacter trogontum in Immunocompetent Patient. Emerging Infectious Diseases. 2016. 22(2) :335.
CrossRef - Abidi M, Wilhelm M, Neff J, Hughes J, Scott C, et al.. Helicobacter canis Bacteremia in a Patient with Fever of Unknown Origin. Journal of clinical microbiology. 51(3) : 1046.
CrossRef - Falsafi T, and Mahboub M. Helicobacter hepaticus, a new pathogenic species of the Helicobacter genus: Similarities and differences with H. pylori. Iranian Journal of Microbiology. 5(3). 185.
- Walker D. Rickestti. International Encyclopedia of Public Health (Second Edition) .2017.
- Gleim D, Kracht M, and Weiss N.. Prokaryotic Nomenclature Up-to-date. Leibniz Institute DSMZ – German Collection of Microorganisms and Cell Cultures.
- Simon J, and Kroneck M. Microbial Sulfite Respiration. Advances in Microbial Physiology. 2013. 62 :4
CrossRef - Kumar M. Chromobacterium violaceum: A rare bacterium isolated from a wound over the scalp. International journal of applied & basic medical research. 2(1): 70.
CrossRef - Sari, E, Etebarian, R, and Aminian H. The effects of Bacillus pumilus, isolated from wheat rhizosphere, on resistance in wheat seedling roots agains the Take-all fungus, Gaeumannomyces graminis var. tritici. Journal of Phytopathology. 2007. 155 :720.
CrossRef - Hernandez J, Bashan, L, Rodriguez D, Rodriguez Y, and Bashan Y. Growth promotion of freshwater microalga Chlorella vulgaris by the nitrogen-fixing, plant growth-promoting bacterium Bacillus pumilus from arid zone soils. European Journal of Soil Biology. 2009. 45:
CrossRef - Huang Y, Li W, Wang L, Lanoot B, Vancanneyt M, Rodriguez C et al. Streptomyces glauciniger sp. nov., a novel mesophilic streptomycete isolated from soil in south China. International Journal of Systematic and Evolutionary Microbiology. 54(6): 2085.
CrossRef - Lim J, Hwan L; Byoung K, and Sunggi H. Draft genome sequence of Pantoea agglomerans R190, a producer of antibiotics against phytopathogens and foodborne pathogens. Journal of Biotechnology. 2014. 188: 7.
CrossRef - Maruyama A, Honda D, Yamamoto H, Kitamura K, and Higashihara T. Phylogenetic analysis of psychrophilic bacteria isolated from the Japan Trench, including a description of the deep-sea species Psychrobacter pacificensis sp. nov. International Journal of Systematic and Evolutionary Microbiology. 2000. 50 (2): 835.
CrossRef - Roinestad F, and Yall I. Volutin granules in Zoogloea ramigera. Appl Microbiol. 1970 19 (6): 973.
CrossRef - Hallberg K, Hedrich S, and Johnson DB. Acidiferrobacter thiooxydans, gen. nov. sp. nov.; an acidophilic, thermo-tolerant, facultatively anaerobic iron- and sulfur-oxidizer of the family Ectothiorhodospiraceae. Extremophiles. 2011. 15(2) :271.
CrossRef - Ionescu D, Bizic M, and Maio N. Community-like genome in single cells of the sulfur bacterium Achromatium oxaliferum . Nat Commun. 8: 455
CrossRef - Grant P, Brenner D, Steigerwalt A, Hollis D, and Weaver R. N. elongata subsp. nitroreducens subsp. nov., Formerly CDC Group M-6, a Gram-Negative Bacterium Associated with Endocarditis. Journal of Clinical Microbiology. 1990: 2591.
CrossRef - Stark B, Dikshit K, and Pagilla K. Recent advances in understanding the structure, function, and biotechnological usefulness of the hemoglobin from the bacterium Vitreoscilla. Biotechnol Lett. 33(9) : 1705.
CrossRef - Emerson D, Reitner J, and Thiel V. Leptothrix. Encyclopedia of Geobiology Springer Dortrecht. 2013.

This work is licensed under a Creative Commons Attribution 4.0 International License.



