

How to Cite | Publication History | PlumX Article Matrix
Rolla Abdalkader Ahmed1,2, Yassir A. Almofti1* and Khoubieb Ali Abd-elrahman3
and Khoubieb Ali Abd-elrahman3
1Department of Molecular Biology and Bioinformatics, College of Veterinary Medicine, University of Bahri, Khartoum- Sudan.
2Department of Microbiology, Faculty of laboratory science, Omdurman Ahlia University, Khartoum- Sudan.
3Department of pharmaceutical technology, College of Pharmacy, University of Medical Science and Technology (MUST) Khartoum- Sudan.
Corresponding Author E-mail: yamofti99@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/2902

ABSTRACT:
Hepatitis B virus (HBV) is the most common cause of hepatocellular carcinoma and liver cirrhosis with significant morbidity and mortality worldwide. DNA polymerase protein of HBV is the immunogenic protein inducing immune response against B and T cells. The aim of this study wasto develop multi-epitope vaccine fromthe polymerase protein elicitingimmune responses.The predicted vaccine comprises epitopes against B and T lymphocytesobtained by IEDB server. The predicted epitopes were linked via suitable spacers (linkers). The 50S ribosomal protein L7/L12 was used as an adjuvant at amino terminal and His-tag at the carboxyl terminal of the vaccine construct. The candidate vaccine contains 457aa and was potentially antigenic and nonallergic. Vaccine molecular weightwas 50.03 KDa with pI of 10.04. The instability index was 25.78 and GRAVY was -0.354 indicating stability andhydrophilicity of the chimeric vaccine,respectively.Vaccine structure (Secondary and tertiary structures) were predicted, refined and used for molecular docking with TLR4.The docking with TLR4 provided energy scores of -1458.7 and -1410.3 for chain A and B, respectively, demonstrated strong binding between the chimeric vaccine and TLR4 chains.The vaccine provided favorable solubility compared to E. coli proteins. Stability via disulfide bonds engineering was predicted to reduce the entropy and mobility regions invaccine construct. Molecular dynamics simulation wasperformed to strengthen the prediction. In silicomolecular cloning was usedto guarantee the efficient clonabilityof the vaccine and translation within suitable vector.
KEYWORDS: B Cells; Chimeric Vaccine; HBV; Immunoinformatics; T Cells
Download this article as:| Copy the following to cite this article: Ahmed R. A, Almofti Y. A, Abd-elrahman K. A. Structural Analysis of the Polymerase Protein for Multiepitopes Vaccine Prediction against Hepatitis B Virus. Biosci Biotech Res Asia 2021;18(1). |
| Copy the following to cite this URL: Ahmed R. A, Almofti Y. A, Abd-elrahman K. A. Structural Analysis of the Polymerase Protein for Multiepitopes Vaccine Prediction against Hepatitis B Virus. Biosci Biotech Res Asia 2021;18(1). Available from: https://bit.ly/3sbeWwv |
Introduction
Hepatitis B virus (HBV) is spherical enveloped double stranded DNA virus (dsDNA), that belongs to the genus Orthohepadnavirusa part of the Hepadnaviridae family [Francki et al., 2012]. Hepatitis caused by HBV can be acute or chronic resulting inhepatocellular carcinoma and liver cirrhosis [Beasley et al., 1981]. It has also been suggested that this virus might cause pancreatitis[Shepard et al., 2006]. HBV is the main cause of viral hepatitis worldwide with chronic carriers exceeding 240 million[Ott et al., 2012]. It is estimated that 1 million of United States citizen at risk of the viral infection specially individuals with sexual behaviors, drug users, healthcare workers, during organ transplantation, individuals with frequent blood transfusions, newborns during parturition and patients with kidney dialysis[Ott et al., 2012]. In addition to that,780,000 people were succumbed to death due to hepatitis B infection worldwide. The most regions that are endemic with HBV are East Asia and sub-Sahara of Africa. In these regions more that 10% of adults were considered as chronic carriers [Yousif et al., 2013]. In developed countries this figure significantly reduced, includingonly less than 1% of the population [Yousif et al., 2013].
Sudan is one of the countries with high sero-prevalence of HBV with 47%–78%people exposed to Hepatitis B surface Antigen (HBsAg). The prevalence varied from about 6.8% to 26% in Central and South Sudan, respectively [Schattner, 2005]. Studies demonstrated that the infection was mainly concentrated in Southern Sudan in early childhood, while the rate of infection was highin Northern Sudanbased on increasing patient’sage. As HBV was recorded asthe main cause of hepatocellular carcinoma and chronic liver disease, in Sudan the disease was recorded as the second main etiology of acute liver failure [Rehermann et al., 1995; Mudawi, 2008].
Few studies demonstrated the prevalence and the risk factors associated with hepatitis B infection in rural Sudan [Rehermann et al., 1995; Mudawi, 2008]. The studies demonstrated that the prevalence of the HBsAg was highest in patients less than five years of age (32.3%). Further more Hepatitis B e Antigen (HBeAg) was recorded in 70% of the pregnant women with HBsAg-positive. The prevalence of the infection in residence with parenteral Malaria therapy was found to be independent. However the studies showed that age, crowding and tattooing were predictive of sero-positivity for hepatitis marker [Mudawi, 2008].
HBV contains many antigens like M glycoprotein, L glycoprotein, S glycoprotein and DNA polymerase protein. Multiple studies showed that DNA polymerase protein is the best protein with immune response against both B and T cells of the immune system [Mudawi, 2008; Bekele et al., 2015; Parkin and Cohen, 2001; Percus et al., 1993]. Polymerase protein is considered as the only immunogene of Hepatitis B virus that induced immune response. Polymerase‐specific cells were present in peripheral blood mono nuclear cells (PBMCs) from the different HBV patients [Mudawi, 2008; Bekele et al., 2015]. Moreover the polymerase includes three domains that differ from each other such asreverse transcriptase (RT), terminal protein (TP) and RNase H. The humoral immune responses related to these proteins is not well clarified, despitethe antibody topolymerase was shown to bepresent in the serum of patients with chronic hepatitis Base [Enshell-Seijffers et al., 2003].
Vaccination against HBV is the effective way to combat the HBV infection. However HBV vaccination such as subunit vaccine-HBsAg demonstrated multiple drawbacks. For instance, three doses are required to achieve full effective course of vaccination. This resulted in difficulties to achieve because of the difficult logistic conditions in some areas and the poor compliance of the patients. In addition to that, there is a comparably high rate, about 5% in adults, considered asnon-responders to the vaccine [Enshell-Seijffers et al., 2003]. Finally, the possibility of some strains of HBV that demonstrate mutations in HBsAg could escape the immunity induced by the present vaccines [Enshell-Seijffers et al., 2003]. Most importantly the association between multiple sclerosis and the recombinant hepatitis B vaccine was become prominent. There was a clear association between hepatitis B recombinant vaccine and the development of the multiple sclerosis in adults that requires multiple precautions [Schattner, 2005]. The vaccination against hepatitis B post liver transplantation in case of hepatitis B–related liver disease was analyzed as an alternative strategy. However this strategy ofreinfection against prophylaxis of hepatitis B immunoglobulin (HBIG) provided conflicting results. In the majority of the studies, HBIG remedy was not continued before vaccination [Potocnakova et al., 2016]. A good significant response via vaccination was achieved under the continuous HBIG injection using hepatitis B surface antigen (HBsAg)‐based vaccine with special adjuvants. The special adjuvants and the continuous HBIG injections were extensively discussed as important factors to enhance good response. However the conventional HBsAg,despite the continued HBIG treatment, the vaccine was potentially incapable in inducing a significant humoral immune response in most treated patients [Potocnakova et al., 2016]. The prepared DNA vaccine to treat hepatitis B virus showed no response or non-sustainable responses compared to the hepatitis B conventional vaccine. Despite this vaccine was shown to be safe with remarkable tolerance and elicited antibodies responses in the vaccinated subjects, failed to induce long lasting immunity[Frikha-Gargouri et al., 2008].
Thus in this regard the need for an efficient, safe vaccine free from the future complications is required. Thus this study attempted to exploit the immunoinformatics approaches to predict epitopes from DNA polymerase protein that provoke the human immune system and to work as safe and effective vaccine against HBV.
Material and Methods
The polymerase protein sequences retrieval
A total of 148Hepatitis B polymerase sequences wereobtained and downloadedfrom NCBI database at (http://www.ncbi.nlm.nih.gov/ protein/polymerase). These quences of the strains were from different countries. The retrieved strains accession numbers, country and the year of collection were presented in table (1).
Table 1: Hepatitis B polymerase retrieved strains with their accession numbers, country and area of collection.
| Accession No | Year | Country | Accession No | Year | Country | Accession No | Year | Country |
| YP_009173866* | NA | USA | ACF95205.1 | NA | Siberia | AFB76718.1 | 1998 | Canada |
| ABD36968 | NA | Eastern India | AGB97500.1 | 1994 | New Zealand | AFB76714.1 | 1998 | Canada |
| AJR19220 | 2003 | Brazil | AGB97371.1 | 1994 | New Zealand | AFB76726.1 | 1998 | Canada |
| AJR19215 | 2003 | Brazil | AGB97364.1 | 1994 | New Zealand | AFB76722.1 | 1998 | Canada |
| ALS87644 | 2007 | South Africans | AGB97322.1 | 1994 | New Zealand | AFB76734.1 | 1998 | Canada |
| AJR19235 | 2003 | Brazil | AGB97315.1 | 1994 | New Zealand | AFB76730.1 | 1998 | Canada |
| AIZ95362 | NA | South Africans | AGB97540.1 | 1994 | New Zealand | AEK67261.1 | 2006 | Iran |
| AIJ49989 | 2005 | South Africans | AGB97506.1 | 1994 | New Zealand | AEK67288. | 2006 | Iran |
| AJR19230 | 2003 | Brazil | AGB97496.1 | 1994 | New Zealand | AFQ36956.1 | 2005 | Latvia” |
| ACF95157 | NA | Siberia | AGB97405.1 | 1994 | New Zealand | AIG21752.1 | 2007 | Belgium |
| ACF95157 | NA | Siberia | AGB97308.1 | 1994 | New Zealand | AIG21738.1 | 2007 | Belgium |
| ACF95157 | NA | Siberia | AGB97506.1 | 1994 | New Zealand | AEK67268.1 | 2006 | Iran |
| AJR19230 | 2003 | Brazil | AGB97357.1 | 1994 | New Zealand | AIG21684.1 | 2007 | Belgium |
| AIW81563 | 2009 | Argentina: | AGB97350.1 | 1994 | New Zealand | AGB97429.1 | 1994 | New Zealand |
| ACF95275 | NA | Siberia | AGB97442.1 | 1994 | New Zealand | AGB97329.1 | 1994 | New Zealand |
| ACF95293 | NA | Siberia | AGB97462.1 | 1994 | New Zealand | AIG21651.1 | 2007 | Belgium |
| ACF95257 | NA | Siberia | AGB97455.1 | 1994 | New Zealand | AIG21664.1 | 2007 | Belgium |
| ACF95224 | NA | Siberia | AGB97564.1 | 1994 | New Zealand | AIG21675.1 | 2007 | Belgium |
| ACF95305 | NA | Siberia | AGB97526.1 | 1994 | New Zealand | ADB03542.1 | 2005 | Indonesia |
| ACF95118. | NA | Siberia | AGB97435.1 | 1994 | New Zealand | AEK66943.1 | 2006 | Iran |
| ACF95251 | NA | Siberia | AGB97378.1 | 1994 | New Zealand | AEK67300.1 | 2006 | Iran |
| ACF95238 | NA | Siberia | AGB97301.1 | 1994 | New Zealand | AEK67241.1 | 2006 | Iran |
| ACF95268 | NA | Siberia | AGB97448.1 | 1994 | New Zealand | AEK67248.1 | 2006 | Iran |
| AGB97469.1 | 1994 | New Zealand | AGB97343.1 | 1994 | New Zealand | AFB76750.1 | 1998 | Canada |
| AFQ36977.1 | 2005 | Latvia | AGB97336.1 | 1994 | New Zealand | AFB76746.1 | 1998 | Canada |
| ACF95261.1 | NA | Siberia | AGB97513.1 | 1994 | New Zealand | AGB97476.1 | 1994 | New Zealand |
| ACF95245.1 | NA | Siberia | AGB97557.1 | 1994 | New Zealand | ABD36978.1 | NA | India |
| AFQ36977.1 | 2005 | Latvia” | AGB97412.1 | 1994 | New Zealand | AGR65533.1 | 2008 | Sudan |
| ACF95331.1 | NA | Siberia | AGB97533.1 | 1994 | New Zealand | AGB97877.1 | 1994 | New Zealand |
| ACF95325.1 | NA | Siberia | AGB97489.1 | 1994 | New Zealand | AGB97613.1 | 1994 | New Zealand |
| ACF95123 | NA | Russia | AGB97520.1 | 1994 | New Zealand | AGB97640.1 | 1994 | New Zealand |
| ACF95311.1 | NA | Siberia | AGB97385.1 | 1994 | New Zealand | AGB97634.1 | 1994 | New Zealand |
| ACF95279.1 | NA | Siberia | AIG21771.1 | 2007 | Belgium | AGB97851.1 | 1994 | New Zealand |
| ACF95133.1 | NA | Siberia | AEK67275.1 | 2006 | Iran | AGB97890.1 | 1994 | New Zealand |
| ACF95128.1 | NA | Siberia | AEK67294.1 | 2006 | Iran | AGB97845.1 | 1994 | New Zealand |
| ACF95338.1 | NA | Siberia | AIG21690.1 | 2007 | Belgium | AGB97870.1 | 1994 | New Zealand |
| ACF95318.1 | NA | Siberia | AIG21698.1 | 2007 | Belgium | AGB97884.1 | 1994 | New Zealand |
| AFQ36970.1 | 2005 | Latvia | AIG21719.1 | 2007 | Belgium | AGB97587.1 | 1994 | New Zealand |
| AFQ36949.1 | 2007 | Latvia | AIG21658.1 | 2007 | Belgium | AGB97654.1 | 1994 | New Zealand |
| ACF95299.1 | NA | Siberia | AIG21681.1 | 2007 | Belgium | AGB97399.1 | 1994 | New Zealand |
| ACF95286.1 | NA | Siberia | AIG21759.1 | 2007 | Belgium | AGB97581.1 | 1994 | New Zealand |
| AGB97728.1 | 2001 | New Zealand | AIG21745.1 | 2007 | Belgium | AGB97593.1 | 1994 | New Zealand |
| AGB97714.1 | 2001 | New Zealand | AIG21765.1 | 2007 | Belgium | AIW68019.1 | NA | Cuba |
| AGB97721.1 | 2001 | New Zealand | AIG21731.1 | 2007 | Belgium | AEK66844.1 | 2006 | Iran |
| AGB97701.1 | 2001 | New Zealand | AGB97707.1 | 1994 | New Zealand | AFB76738.1 | 1998 | Canada |
| ACF95212.1 | NA | Siberia | AEK67255.1 | 2006 | Iran | ACH58048.1 | 2005 | China |
| ACF95231.1 | NA | Siberia | AIG21705.1 | 2007 | Belgium | AFB76742.1 | 1998 | Canada |
| ACF95345.1 | NA | Siberia | AGB97482.1 | 1994 | New Zealand | ACF95218.1 | NA | Siberia |
| AGB97857.1 | 1994 | New Zealand | AEK67282.1 | 2006 | Iran | ACF95152.1 | NA | Russia |
| AGB97885.1 | 1994 | New Zealand | – | – | – | – | – | – |
*Ref sequence
N/A: not available
Multiple sequences alignment and epitopes conservancy
Multiple sequence alignment was used to align the strains sequences usingthe offline BioEdit program, version 7.0.9.0[Hall 1999]. Epitopes that demonstrated 100% conservancy from the aligned sequences of the variant HBV strains were obtained by finding their positions in the sequences with no mutation. The conserved epitopes were further subjected and analyzed by the free Immune Epitope Database prediction resources (IEDB)at (http://www.iedb.org/)
B cell epitopes prediction
Epitope is a discrete part of the antigen recognized by the immune system, particularly, B and T cells and to which an antibodies bind. Antibodies bound to epitopes viaparatopes that formed by continuous sequences of particular amino acids from the antigen[Percus et al., 1993]. B cells are subtypes of white blood cells known as lymphocyte sub type. They function as humoral immunity component of the adaptive immune system by secreting antibodies[Parkin and Cohen, 2001]. Additionally, B cells present antigen (they are also classified as professional antigen-presenting cells (APCs) and secrete cytokines [Parkin and Cohen, 2001].The IEDB resource tool at (http://toolsiedb.ofg/bcell/) was used to predict B cell epitopes. Eg; BepiPred was used to predict the linearB-cell epitopes [Larsen et al., 2006; Ponomarenko and Bourne, 2007; Haste Andersen et al., 2006]. Antigen surface epitopes were predicted by Emini surfaceaccessibility tools[Emini et al., 1985]. Kolaskar and Tongaonker antigenicity method was used to determine the antigenicity of the predicted epitopes [Kolaskar and Tongaonkar, 1990].The threshold of each prediction tool was obtained by IEDB that calculated the threshold as the average prediction score for each corresponding tool.
T cells epitopes prediction
T lymphocyte plays a central role in cell-mediated immunity. They functioned by recognition of major histocompatibility complex molecules (MHC) in a set of surface proteins in antigen presenting cells. The MHC is important for the immune system to recognize foreign molecules in vertebrates and determines their histocompatibility [Janeway, 1989].
Cytotoxic T lymphocytes epitopes prediction
IEDB MHC I prediction tool at (http://tools.iedb.org/mhci/) was used to analyze the peptides bound to MHC I molecules. The prediction undergoesmultiple steps; the prediction of the cleaved epitopes that boundto MHC groove, followed by choosing ArtificialNeural Network (ANN) as a prediction method. Before prediction initiated, the epitopes lengths were adjusted as9mers. The half-maximal inhibitory concentration (IC50) wasset to be ≤300 [Kim et al., 2012; Nielsen et al., 2003; Lundegaard et al., 2008; Sidney et al., 2008].
Helper T lymphocytes epitopes prediction
IEDB MHC II prediction tool at (http://tools.immuneepitope.org/mhcii/) was used to analyzethe peptide bound to MHC class II molecules. A set of reference alleles was used to predict the binding of epitopes toMHC II groove with different lengths. Such binding variability features makes prediction process difficult with low accuracy. The prediction methodused was NN- align that uses the artificial neural networks that permits simultaneous identification of epitopes bound to MHC II with high binding affinity. The half-maximal inhibitory concentration (IC50) was set to be ≤3000[Wang et al., 2008].
Antigenicity, allergenicity and toxicity of the predicted epitopes
VaxiJen v2.0 server at (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was used to investigate the antigenicity of the predicted epitopes using the default threshold of the server (0.4). AllerTOP server at (http://www.ddg-pharmfac.net/AllerTop/)[Dimitrov et al., 2013]and Toxin Pred server at (http://crdd.osdd.net/raghava/toxinpred/) [Gupta et al., 2013] werethe methods used to predict the allergenicity and toxicity of the epitopes interacting with B and T lymphocytes, respectively.
Calculation of the population coverage
IEDB tool at (http://tools.iedb.org/tools/population/iedb_input) for calculation of the population coverage was used to calculate the population coverage for each T lymphocytes predicted epitopes. The prediction of theMHC I and MHC II potential binders from polymerase protein was determined against the whole world.
Assemblage of the multi-epitope vaccine
Antigenic, nonallergic and nontoxic epitopes that interacted with T helper, T cytotoxic and B lymphocytes were used to generate vaccine construct. The T cytotoxic epitopes were fused the GPGPG linker [Shey et al., 2019] while the B and T helper cells epitopes were fused with KK linker [Hasan et al., 2019]. Insertion of linkers between two epitopes provides efficient separation that is required for the efficient functioning of each epitope [Nezafat et al., 2014; Ali et al., 2017]. The 50S ribosomal protein L7/L12 of Mycobacterium tuberculosis (strain ATCC 25618/ H37Rv, with uniprot accession no P9WHE3)was used as an adjuvant on the amino terminal of the vaccine sequence to enhance the immunogenicity of the chimeric vaccine. The adjuvant was joined via EAAAK linker to the epitopes. Six His-tags were added at the carboxyl terminal for purification and identification of the chimeric vaccine.
Physical and chemical characterisitics of the chimeric vaccine
ProtParam server at (https://web.expasy.org/protparam/) was used to analyze the physical and chemical properties of the chimeric vaccine. The calculated features comprises: molecular weight (MW), theoretical isoelectric point (pI), atomic composition, amino acid composition, extinction coefficient, estimated half-life, aliphatic index, instability index and grand average of hydropathicity (GRAVY)
Secondary structure prediction
Self-optimized prediction method (SOPMA) at (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html) was used to predict the secondary structure of the multi-epitope vaccine protein [Combet et al., 2000].
Tertiary structure prediction
The tertiary structure of final multi-epitope vaccine was predicted by submitting the sequence of the chimeric vaccine sequence to PHYRE-2 protein folding recognition server (http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index ) [Kelley et al., 2015]. The server used to analyze and predict the3D structure of the protein, functionsand mutations. The output PDB file obtained was used for refinement and adaptation of the chimeric vaccine structure.
Tertiary structure refinement and validation
The PDB file obtained by PHYRE-2 server was submitted to Galaxy WEB web server for protein structure prediction and refinement [Shin et al., 2014; Ko et al., 2012]. Template based refinement model of the 3D tertiary structure was performed to ameliorate both the local and global structural quality. Thestructure validation was performed through Ramachandran plot at RAMPAGE [Al-Hakim et al., 2015; Lovell et al., 2002]. Moreover the PDB file obtained by PHYRE-2 server was analyzed by ProSA server for structure potential errors [Wiederstein and Sippl, 2007].
Prediction of discontinuous B-cell epitopes
The prediction of discontinuous B-cell epitopes was performed using ElliPro in theIEDB at (http://tools.iedb.org/ellipro/ ).ElliProtool predicts discontinuous and linear antibody epitopes depending on the protein (antigen) 3D structure. ElliProcombines each of the predicted epitope with a score, known as Protrusion Index (PI) value, averaged over the epitope residues. The minimum score and the maximum distance (Angstrom) of the prediction were set to default (0.5) and (6), respectively [Ponomarenko et al., 2008].
Solubility of the chimeric vaccine
The solubility of the chimeric protein was measured compared to the proteins of the E. coli using protein-sol server at (https://protein-sol.manchester.ac.uk/). Protein sol is a web based suite of theoretical calculations and predictive algorithms for understanding protein solubility of a given protein (QuerySol, scaled solubility value) compared to the E. coli experimental dataset (PopAvrSol) with a population average of 0.45[Hebditch et al., 2017]. Protein scored greater than 0.45 is expected to be soluble compared to the average solubility of E. coli proteins[Hebditch et al., 2017; Niwa et al., 2009].
Stability of the chimeric vaccine
The geometric conformation made by disulfide bonds engineering strengthens the chimeric vaccine with significant stability. The Disulfide by Design 2.0 (DbD2) is a web-based tool for disulfide engineering in proteins was used to engineer disulfide bonds in the chimeric vaccine [Craig and Dombkowski, 2013]. The position of the predicted disulfide bonds for residue pairs in the protein located in the high mobile regions. It is calculated based on the chi3 residue screening, B-factor value and energy value (equal to or less that 3.5), assuming the residue pairs mutated to cysteine.
Molecular dynamics simulation
iMODS is an online server (http://imods.chaconlab.org/) explores the collective motions of proteins and nucleic acids using normal modes analysis (NMA) in internal coordinates[Lopez-Blanco et al., 2014]. The server was used to analyze the stability of protein-protein complex and further effectively assess the structural dynamics of protein complex [Prabhakar et al., 2016; Awan et al., 2017]. The iMODS provided the direction and magnitude of the motions in protein complex in the form of deformability, eigenvalues, B-factors, covariance, variance map in the residue index and elastic network in the atoms index [Lopez-Blanco et al., 2011].
Molecular docking of the chimeric vaccine with TLR4
The automated docking server,ClusPro 2.0, that uses the discrimination method for prediction of protein complexeswas used for molecular interaction between the vaccine protein and Toll like Receptor 4 (TLR4) [Kozakov et al., 2017; Vajda et al., 2017]. ClusPro 2.0server rapidly filters the docked conformations and ranks them according to their clustering properties. The chimeric vaccine construct PDB file was submitted to the server with TLR4 (PDB4G8A) as a receptor. The docking process was performed in TLR4 chain A and chain B separately. The advance method was used as a docking method.The interaction between the vaccine and TLR4 chains was visualize by thePyMOL visualization tool.
In silico cloning
The insilico cloning was performed to guarantee the expression of the chimeric vaccine in the selected host. The protein of the chimeric vaccine was reversed translated to DNA sequence using Java Codon Adaptation Tool (JCAT) server (http://www.prodoric.de/JCat ). The best codon adaptation index (CAI) score is 1.0 but >0.8 is considered a good score [Morla et al., 2016]. The favourable GC content ranged between 30–70%. The Ndel and Xho1restriction enzymes cutting sites sequences were added to the ends of theDNA sequence. The sequence was inserted into pET28a (+) vector between the Ndel and Xho1 restriction enzymes using SnapGene restriction cloning module[Shey et al., 2019; Pandey et al., 2018].
Results
Epitopes conservancy
Sequence alignment of all retrieved strains of polymerase proteins were performed using ClustalW that presented by Bioedit software. Sequence alignment was performed to obtain 100% conserved epitopes from the retrieved strains. Epitopes conservancy assessed between the reference sequence and all the retrieved sequences via alignment. As shown in figure (1) the retrieved sequences of the polymerases showed conservancy upon aligned. The identity of amino acidswithin the sequences clearly identified the conserved regions.
 |
Figure 1: Multiple sequence alignment showed the conservancy between sequences of the retrieved strains of the polymerase protein. Dots showed the conserved regions while letters within rectangles showed the mutated or the unconserved region between strains |
B-cell Epitopes Prediction
IEDB server was used to predict B–cell epitopes. As shown in figure (2) scores equal to or greater than the thresholds of 0.06 (for linear epitope), 1.000 (for surface accessible epitopes) and 1.049(for antigenic epitopes)were considered as potential epitopes determinants of B cell.The three tools predicted 76 linear conserved epitopes, 46 epitopes on the surface and 30 antigenic epitopes.However only 14 epitopes overlapped the three tools and were further investigated for antigenicity using Vaxijen software with default threshold (0.4), allergenicity and toxicity. Upon investigation only one epitope was shown to be antigenic, nonallergic and nontoxic.This epitope was provided in table (2).
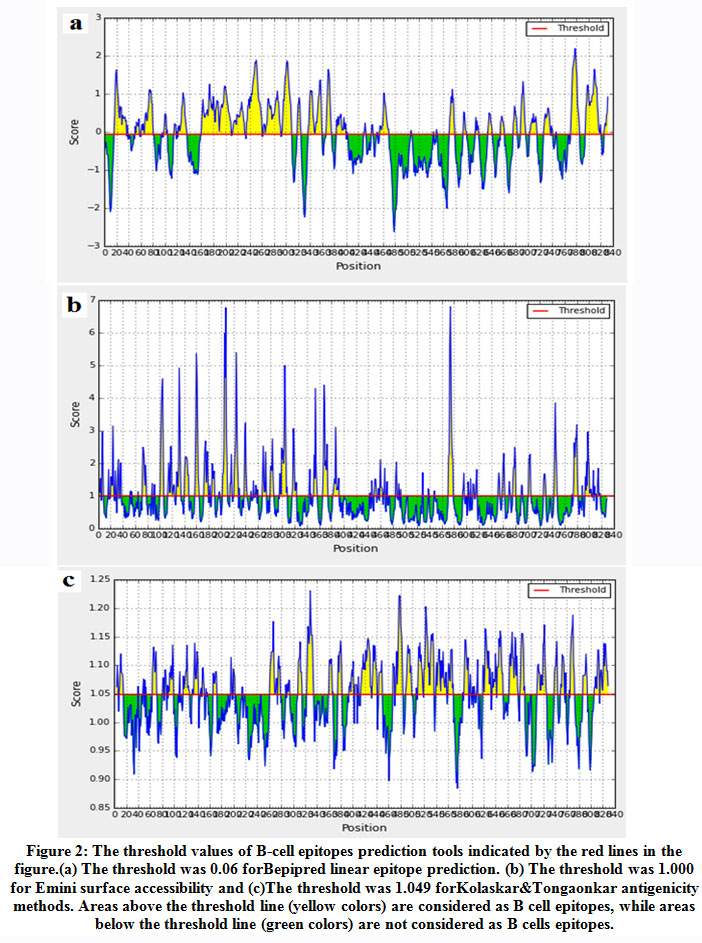 |
Figure 2: The threshold values of B-cell epitopes prediction tools indicated by the red lines in the figure.(a) The threshold was 0.06 forBepipred linear epitope prediction. (b) The threshold was 1.000 for Emini surface accessibility and (c)The threshold was 1.049 forKolaskar&Tongaonkar antigenicity methods. Areas above the threshold line (yellow colors) are considered as B cell epitopes, while areas below the threshold line (green colors) are not considered as B cells epitopes. |
Table 2: Only one epitope predicted against B cell. The length and the threshold of each IEDB prediction tools were shown.
| Epitope | Start | End | Emini
Surface accessibility (1.000) |
Kolaskar and Tongaonkar antigenicity (1.049) | Vaxijen
antigenicity (0.4) |
Allergenicity | Toxicity |
| YLPLDKGIKPYY | 122 | 133 | 1.923 | 1.072 | 0.6637 | non-allergen | Non-Toxin |
Cytotoxic T lymphocytes epitopes prediction
Based on the ANN prediction methodwith IC50≤300,only 22 epitopes were found interacting with different MHC1alleles.Among the 22 epitopes only 7 epitopes were found antigenic, nonallergic and nontoxic. The 7 epitopes and their population coverage scores provided in table (3) and wereelected as MHC-1 epitopes.
Table 3: The 7 predicted epitopes interacted against cytotoxic T cells. The Population coverage against whole world was shown for each epitope.
| Peptide | Start | End | Vaxijin
antigenicity (0.4) |
Allergenicity | Toxicity | Population coverage |
| YFQTRHYLH | 141 | 149 | 0.6793 | non-allergen | Non-Toxin | 9.14% |
| SLDVSAAFY | 416 | 424 | 0.8683 | non-allergen | Non-Toxin | 23.03% |
| QRIVGLLGF | 623 | 631 | 0.5111 | non-allergen | Non-Toxin | 4.78% |
| AELLAACFA | 717 | 725 | 0.514 | non-allergen | Non-Toxin | 3.45% |
| LAACFARSR | 720 | 728 | 1.0703 | non-allergen | Non-Toxin | 5.83% |
| DNSVVLSRK | 737 | 745 | 1.3699 | non-allergen | Non-Toxin | 5.83% |
| RVHFASPLH | 818 | 826 | 0.4556 | non-allergen | Non-Toxin | 3.89% |
Helper T lymphocytes epitopes prediction
Based on NN-align method with IC50 ≤ 3000, a total of 77 predictedepitopes interacted against MHC-II alleles.Among them, only 19 epitopes were antigenic, nonallergic and nontoxic. Table (4) provided the 19 epitopes with their population coverage scores.
Table 4: The 19 predicted epitopes interacted against helper T cells. The Population coverage against whole world was shown for each epitope.
| Core epitope | Peptide | Start | End | Vaxijen antigenicity (0.4) | Allergenicity | Toxicity | Population coverage |
| CSVVRRAFP | CSVVRRAFPHCLAFS | 523 | 537 | 0.4739 | Non-allergen | Non-Toxin | 76.04% |
| DATPTGWGL | DATPTGWGLVMGHQR | 689 | 703 | 2.0429 | Non-allergen | Non-Toxin | 11.53% |
| ESRLVVDFS | ESRLVVDFSQFSRGN | 374 | 388 | 0.511 | Non-allergen | Non-Toxin | 56.72% |
| KTKRWGYSL | KTKRWGYSLNFMGYV | 574 | 588 | 0.9557 | Non-allergen | Non-Toxin | 20.51% |
| KYLPLDKGI | KYLPLDKGIKPYYPE | 121 | 135 | 0.806 | Non-allergen | Non-Toxin | 48.66% |
| LAACFARSR | LAACFARSRSGANII | 720 | 734 | 1.0703 | Non-allergen | Non-Toxin | 52.28% |
| LDKGIKPYY | LDKGIKPYYPEHLVN | 125 | 139 | 0.6475 | Non-allergen | Non-Toxin | 77.23% |
| LDVSAAFYH | LDVSAAFYHLPLHPA | 417 | 431 | 0.906 | Non-allergen | Non-Toxin | 89.97% |
| LGFAAPFTQ | LGFAAPFTQCGYPAL | 629 | 643 | 0.6846 | Non-allergen | Non-Toxin | 91.70% |
| LPLDKGIKP | LPLDKGIKPYYPEHL | 123 | 137 | 0.8282 | Non-allergen | Non-Toxin | 32.01% |
| NSVVLSRKY | NSVVLSRKYTSFPWL | 738 | 752 | 1.2913 | Non-allergen | Non-Toxin | 27.73% |
| NWILRGTSF | NWILRGTSFVYVPSA | 758 | 772 | 1.3089 | Non-allergen | Non-Toxin | 78.80% |
| SVVLSRKYT | SVVLSRKYTSFPWLL | 739 | 753 | 1.4231 | Non-allergen | Non-Toxin | 27.90% |
| TAELLAACF | TAELLAACFARSRSG | 716 | 730 | 0.5238 | Non-allergen | Non-Toxin | 88.45% |
| VHFASPLHV | DRVHFASPLHVAWRP | 817 | 831 | 0.4149 | Non-allergen | Non-Toxin | 99.83% |
| VNHYFQTRH | VNHYFQTRHYLHTLW | 138 | 152 | 0.6801 | Non-allergen | Non-Toxin | 77.52% |
| VVLSRKYTS | VVLSRKYTSFPWLLG | 740 | 754 | 1.194 | Non-allergen | Non-Toxin | 87.65% |
| WKVCQRIVG | WKVCQRIVGLLGFAA | 619 | 633 | 0.7487 | Non-allergen | Non-Toxin | 63.93% |
| YLPLDKGIK | YLPLDKGIKPYYPEH | 122 | 136 | 0.5373 | Non-allergen | Non-Toxin | 55.74% |
Construction of muli-epitopes vaccine (chimeric vaccine)
The chimeric vaccine includes the B cell and T cell predicted epitopes. One epitope was proposed as B cell epitope, seven epitopes as cytotoxic T cell and nineteen epitopes as helper T cell. The chimeric vaccine composed of 457 amino acids after addition of the adjuvants, linkers and 6-His- tag (figure 3). The chimeric vaccine demonstrated antigenicity in Vaxigen server with score of 0.5110 and was nonallergen in the Allertop server.
 |
Figure 3: The chimeric vaccine structure. T helper epitopes (purple color) and B cell epitopes (red color) were linked by the short peptide linker KK, while T cytotoxic epitopes (blue color) were linked by GPGPG linker. The adjuvant (green color) was at amino terminal and the 6-his-tag at the carboxyl terminal. |
Physical and chemical characteristics of the vaccine construct
The MW of the chimeric vaccine was 50.03 KDa with pI value of 10.04. The negatively and positively charged residues in the vaccine structure were 39 and 92 respectively. The Extinction coefficient was 46215 indicating all pairs of Cys residues form cystines. The estimated half-life was 30 hours (mammalian reticulocytes, in vitro), >20 hours (yeast, in vivo) and >10 hours (Escherichia coli, in vivo). The instability index (II) was computed to be 25.78 indicating the stability of the chimeric vaccine. Aliphatic index was 81.82 and the GRAVY was -0.354 indicating the hydrophilicity of the chimeric vaccine.
Secondary structure prediction
Figure (4) demonstrated that Self-optimized prediction method (SOPMA) provided that among the 457 amino acids of the chimeric vaccine 164 amino acid (35.89%) involved in formation of alpha helices, 83 amino acids (18.16%) were extended strands, 43 amino acids (9.41%) were beta turns while 167 amino acids (36.54%) were random coils with no unambiguous or any other states.
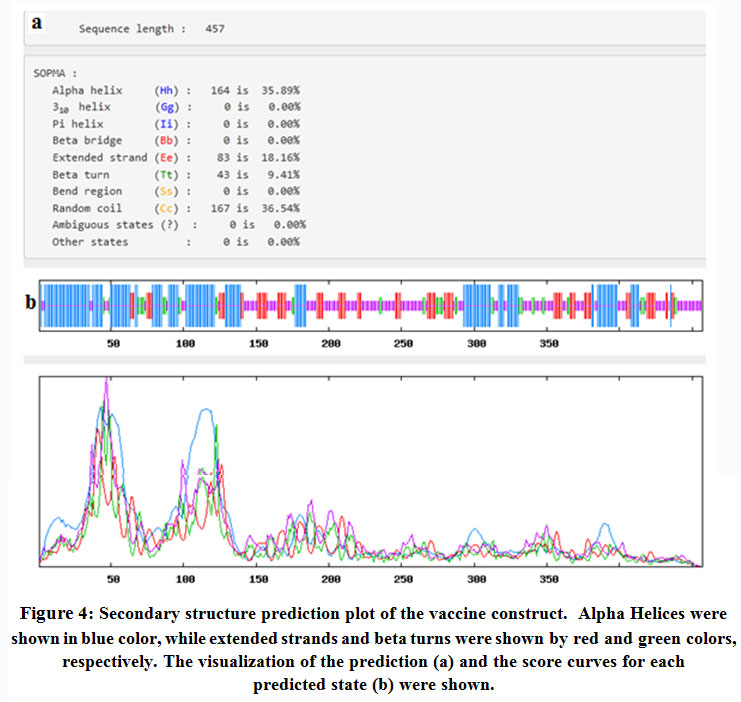 |
Figure 4: Secondary structure prediction plot of the vaccine construct. Alpha Helices were shown in blue color, while extended strands and beta turns were shown by red and green colors, respectively. The visualization of the prediction (a) and the score curves for each predicted state (b) were shown. |
Tertiary structure prediction
Figure (5) provided the 3D structure of the chimeric vaccine predicted by PHYRE2 server.The 3D structure was refined with Galaxyrefine server. The model was further assessed by Ramachandran plot after refinement and demonstrated that the number of residues in favoured, allowed and outlier region were 90.1%, 7.3% and 2.6%,respectively. Moreover proSA server Z-score of the chimeric vaccine was -3.69 which represents the good quality of the model.
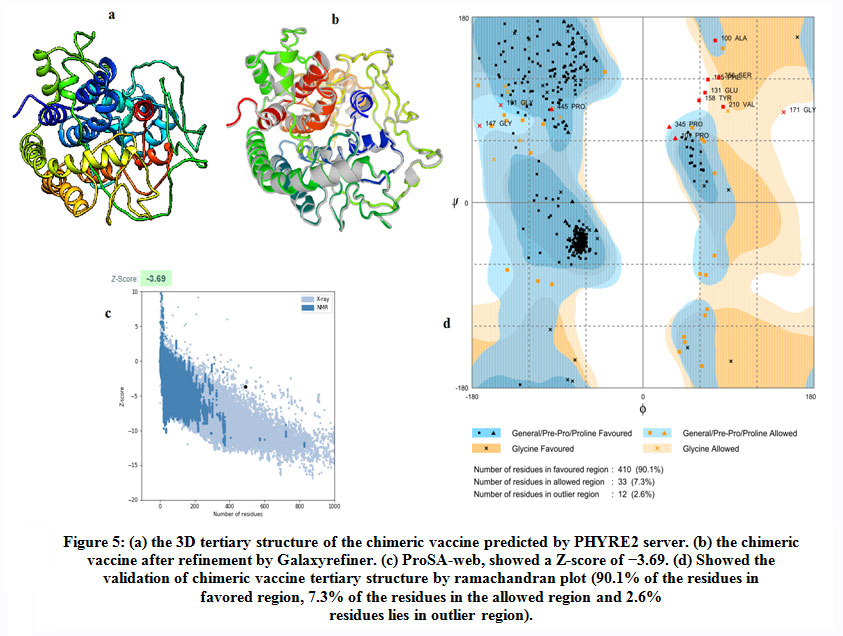 |
Figure 5: (a) the 3D tertiary structure of the chimeric vaccine predicted by PHYRE2 server. (b) the chimeric vaccine after refinement by Galaxyrefiner. (c) ProSA-web, showed a Z-score of −3.69. (d) Showed the validation of chimeric vaccine tertiary structure by ramachandran plot (90.1% of the residues in favored region, 7.3% of the residues in the allowed region and 2.6% residues lies in outlier region). |
Discontinuous B-cell epitopes prediction
Table (5) and figure (6) demonstrated 12 the B-cell discontinuous epitopes. The scores of these epitopes were ranged from 0.558 to 0.769. The total of residues predicted locating in these discontinuous epitopes was 251 residues. The size of the conformational epitopes ranged from 4 to 119 residues.
Table 5: The showed the number of the predicted discontinuous B cell epitopes with the number of the residues and their scores.
| No. | Residues | Number of residues | Score |
| 1 | _:E390, _:A394, _:C395, _:K397, _:K398, _:V399, _:H400 | 7 | 0.769 |
| 2 | _:A251, _:F252, _:P253, _:K254, _:K255, _:D256, _:A257, _:T258, _:P259, _:T260, _:G261, _:W262, _:V271, _:V272, _:D273, _:F274, _:S275, _:K276, _:K277, _:K278, _:T279, _:K280 | 22 | 0.754 |
| 3 | _:F63, _:L67, _:E68, _:A69, _:A70, _:G71, _:D72, _:K73, _:K74, _:I75, _:G76, _:V77, _:I78, _:K79, _:V80, _:E83, _:K94, _:D95, _:V97, _:D98, _:G99, _:A100, _:P101, _:K102, _:A109, _:K110, _:E111, _:D114, _:E115, _:K117, _:A118, _:K119, _:L120, _:E121, _:A122, _:A123, _:G124, _:A125, _:T126, _:V127, _:T128, _:L143 | 42 | 0.703 |
| 4 | _:G159, _:P160, _:G161, _:P162, _:G163, _:Q164, _:R165, _:I166, _:V167, _:G168, _:L169, _:L170, _:G171, _:F172, _:G173, _:P174, _:G175, _:P176, _:G177, _:A178, _:E179, _:L180, _:L181, _:A182, _:A183, _:C184, _:F185, _:A186, _:G187, _:P188, _:G189, _:P190, _:G191, _:L192, _:A193, _:A194, _:C195, _:F196, _:A197, _:S199, _:D206, _:N207, _:S208, _:V209, _:V210, _:L211, _:S212, _:R213, _:K214, _:G215, _:P216, _:G217, _:P218, _:G219, _:R220, _:V221, _:H222, _:F223, _:A224, _:S225, _:P226, _:L227, _:H228, _:K229, _:K230, _:L232, _:D312, _:K313, _:G314, _:I315, _:K316, _:P317, _:Y318, _:Y319, _:K320, _:K321, _:L322, _:D323, _:V324, _:S325, _:A326, _:A327, _:F328, _:Y329, _:H330, _:K331, _:K332, _:L333, _:G334, _:F335, _:A336, _:A337, _:P338, _:F339, _:K342, _:K343, _:L344, _:P345, _:L346, _:K348, _:G349, _:I350, _:K351, _:P352, _:K353, _:K354, _:N355, _:S356, _:R370, _:G371, _:T372, _:S373, _:F374, _:K375, _:K376, _:S424, _:K426, _:Y427, _:H457 | 119 | 0.701 |
| 5 | _:E131, _:A132, _:A133, _:A134, _:K135, _:Y136, _:F137, _:Q138, _:T139, _:Y142, _:F401, _:A402, _:S403, _:P404, _:L405, _:H406 | 16 | 0.663 |
| 6 | _:A302, _:C303, _:A305, _:R306, _:S307, _:K309, _:K310 | 7 | 0.659 |
| 7 | _:G263, _:L264, _:K265, _:K266, _:E267, _:L270 | 6 | 0.624 |
| 8 | _:E59, _:Y290, _:L291, _:P292, _:L293, _:D294, _:K298 | 7 | 0.605 |
| 9 | _:M1, _:A2, _:K3, _:L4, _:S5, _:E8, _:F32, _:E33, _:V34, _:S246, _:R249, _:R250 | 12 | 0.602 |
| 10 | _:V378, _:V379, _:K383 | 3 | 0.587 |
| 11 | _:R82, _:G87, _:L88, _:G89, _:L90, _:K91 | 6 | 0.56 |
| 12 | _:G283, _:L286, _:K287, _:K289 | 4 | 0.558 |
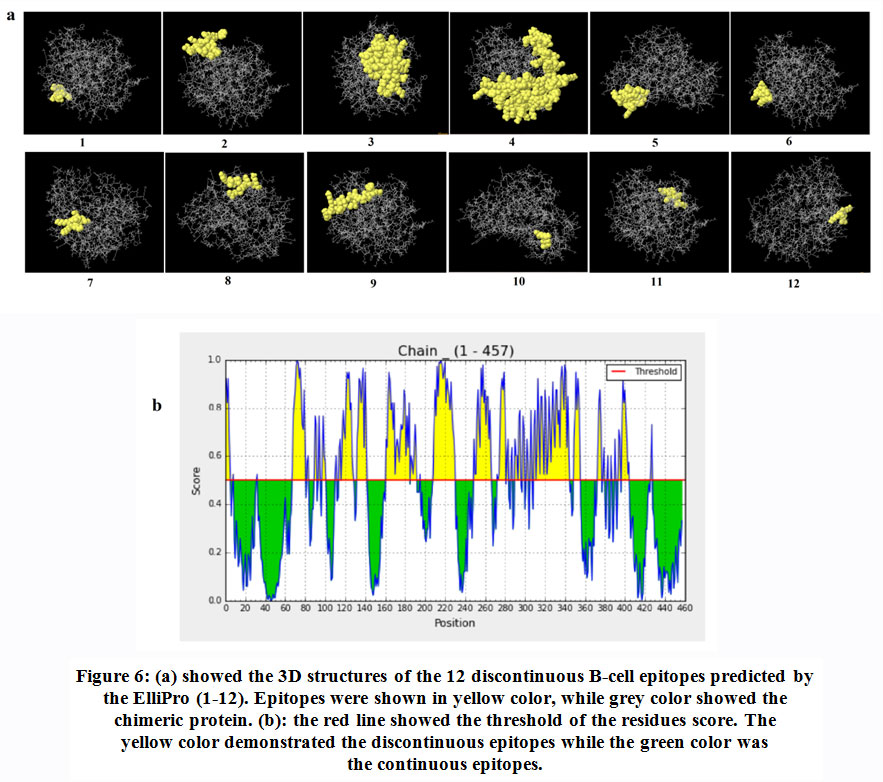 |
Figure 6: (a) showed the 3D structures of the 12 discontinuous B-cell epitopes predicted by the ElliPro (1-12). Epitopes were shown in yellow color, while grey color showed the chimeric protein. (b): the red line showed the threshold of the residues score. The yellow color demonstrated the discontinuous epitopes while the green color was the continuous epitopes. |
Solubility of the chimeric vaccine
Figure (7) showed the solubility of the chimeric vaccine, QuerySol scaled solubility value, was 0.567 compared to the experimental data set (PopAvrSol) of 0.45 for E. coli proteins. This result showed that the chimeric vaccine is potentially soluble.
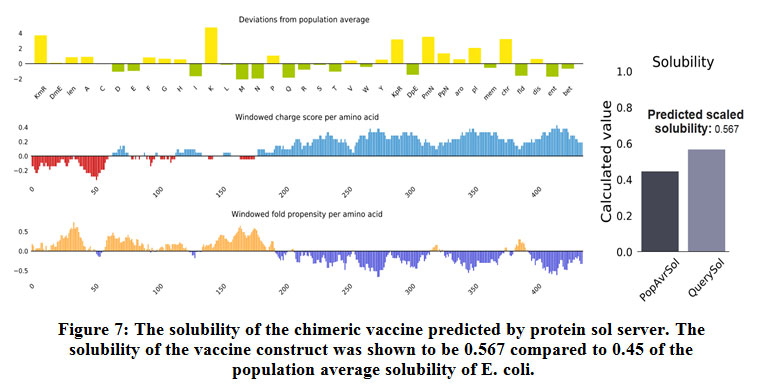 |
Figure 7: The solubility of the chimeric vaccine predicted by protein sol server. The solubility of the vaccine construct was shown to be 0.567 compared to 0.45 of the population average solubility of E. coli. |
Stability of the chimeric vaccine
Figure (8), showed the disulfide engineered residues in the chimeric protein sequence when mutated to cysteine. A total 36 pairs of amino acids residues probably forming disulfide bond. But only five regions were evaluated to form disulfide bond based on the chi3 residue screening (between −87 and +97), B-factor value (ranged 6.950 – 17.410) and energy value less than 3.5. The five residue pairs were 63PHE-113ALA; 147GLY-443TYR; 219GLY-333LEU; 277LYS-280LYS and 293LEU-296GLY.
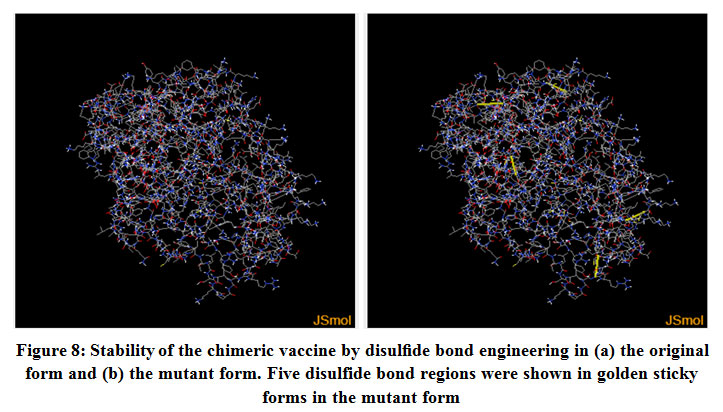 |
Figure 8: Stability of the chimeric vaccine by disulfide bond engineering in (a) the original form and (b) the mutant form. Five disulfide bond regions were shown in golden sticky forms in the mutant form |
Molecular dynamics simulation
The stability and the large scale mobility of chimeric protein were shown in figure (9) that performed by NMA (Normal mode analysis) in the iMODS server. The direction of the mobility of each residue in the chimeric vaccine protein was indicated by arrows(figure 9-a). Moreover the deformability of the molecule was associated with the distortion of the individual residues. This was indicated by hinges in the chain (figure 9-b). The experimental B-factor was obtained from the corresponding PDB field and calculated from NMA(figure 9-c). The eigenvalue which represents the motion stiffness was calculated to be 2.142287e−04 (figure 9-d), where the least the eigenvalue, the easier the deformation. Figure (9-e)provided the covariance matrix that represents the coupling between pairs of residues, i.e. whether they experience correlated motion (red color), uncorrelated motion (white color) or anti-correlated motion (blue color). The elastic network model defines which pairs of atoms are joined by springs in which each dot represents one spring between the corresponding pair of atoms. Dots are colored based on their stiffness. The darker grays represents stiffer springs and vice versa (figure 9-f).
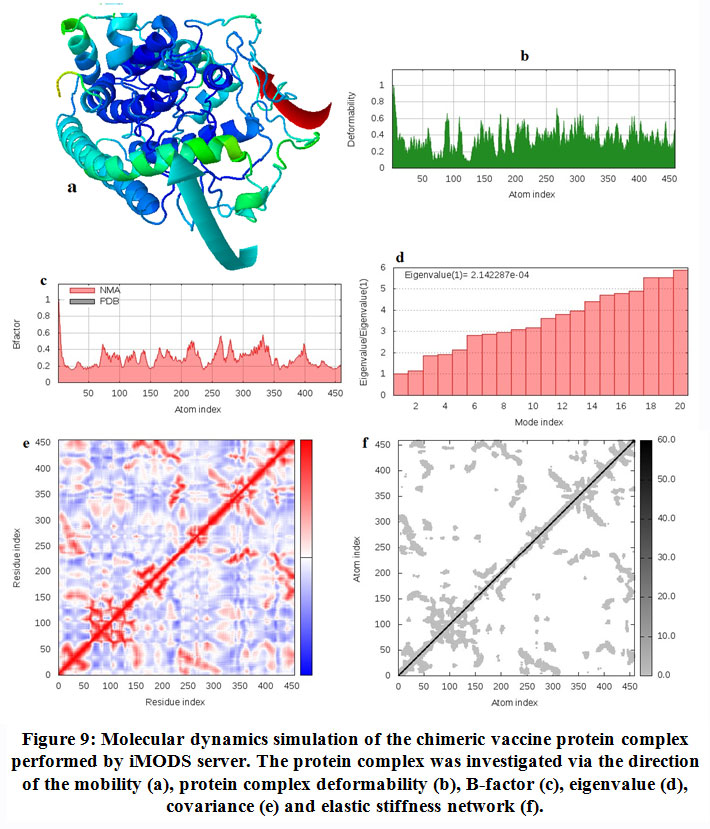 |
Figure 9. Molecular dynamics simulation of the chimeric vaccine protein complex performed by iMODS server. The protein complex was investigated via the direction of the mobility (a), protein complex deformability (b), B-factor (c), eigenvalue (d), covariance (e) and elastic stiffness network (f). |
Molecular docking of the chimeric vaccine with TLR4
Molecular docking was performed between the TLR4 and the chimeric vaccine protein and provided biologically significant results indicated by the terms of free binding energy. The docking process for TLR4 chain A provided that the representative lower energy score was-1458.7 (figure 10-a). For chain B the representative lower energy score was -1410.3 (figure 10-b).These negatively scored values demonstrated the strong binding between the chimeric vaccine protein and TLR4 chains.
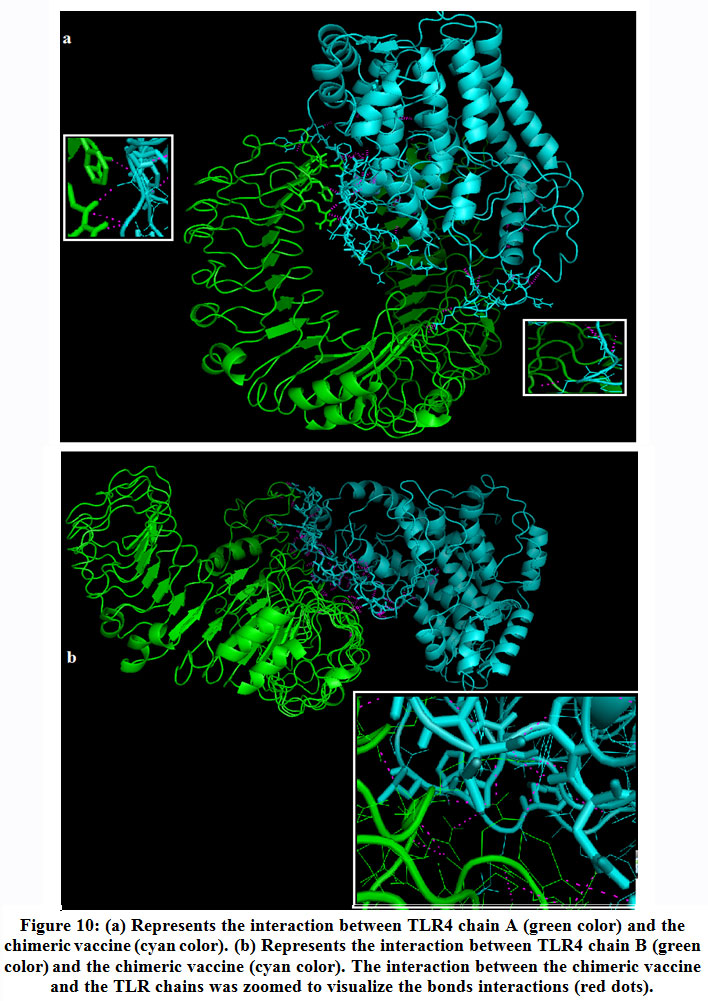 |
Figure 10: (a) Represents the interaction between TLR4 chain A (green color) and the chimeric vaccine (cyan color). (b) Represents the interaction between TLR4 chain B (green color) and the chimeric vaccine (cyan color). The interaction between the chimeric vaccine and the TLR chains was zoomed to visualize the bonds interactions (red dots). |
In silico cloning of the chimeric vaccine
The DNA sequence of the chimeric protein provided CAI-Value of 0.9199, representing the higher proportion of most abundant codons. While the GC-content was 51.58%, indicating favourable GC content. Figure (11), showed that DNA sequence was cloned into pET28a (+) vector betweenNdel and Xho1restriction enzymes cutting sites.
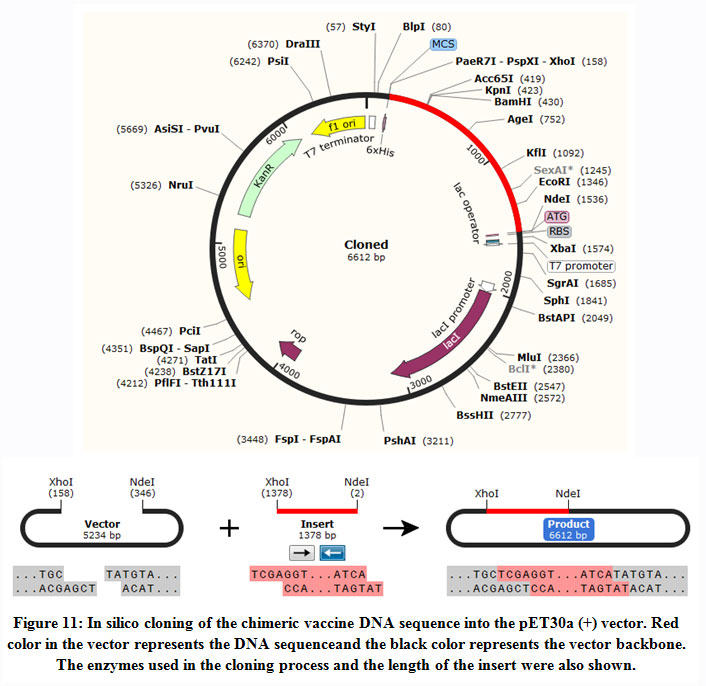 |
Figure 11: In silico cloning of the chimeric vaccine DNA sequence into the pET30a (+) vector. Red color in the vector represents the DNA sequenceand the black color represents the vector backbone. The enzymes used in the cloning process and the length of the insert were also shown. |
Discussion
HBV infection includes large spectrum of hepatic diseases ranged from acute to chronic hepatitis, hepatocellular carcinoma (HCC) and liver cirrhosis [Hollinger and Liang, 2001]. Multiple novel remedies against HBV were reported. For instance the Direct Acting Anti-virals (DAA) directly targeted the virus via multiple inhibition events. This includes entry inhibitors, site-specific cleavage of DNA agents, polymerase inhibitors, inhibitors of relax-circular DNA to covalently closed circular DNA (cccDNA) conversion. Inhibition events also includes the inhibitors of nucleocapsid assembly, agents that knockdown HBV RNA, viral proteins and HBV DNA knockdown, capsid inhibitors and agents that block HBsAg secretion [Ward et al., 2016; Zheng et al., 2017]. Moreover, beside the inhibition events, the host targeting agents (HTA) acted by improving the ability of the host immunity. This includes inhibitors of the immune checkpoints, stimulators of exogenous interferon, therapeutic vaccines and agents that enhances APOBEC3A and APOBEC3B [Ward et al., 2016; Zheng et al., 2017; Lin and Kao, 2016; Jia et al., 2015].The advances made in the field of the recombinant DNA technology guided to the development of second-generation recombinant vaccines production against HBV in yeast [Sitrin et al., 1993; McMahon et al., 1992; McAleer et al., 1984].Althoughthe second-generation HBV vaccines shown to be as an efficient vaccine, the immunization failure might occur and can be explained by variable factors such as the non-responsiveness to immunization that might be genetically determined resistance [Craven et al., 1986; Alper et al., 1989]. Moreover the ability to produce antibodies in response to immunization is controlled by autosomal dominantly expressed HLA class II molecules [Craven et al., 1986; Alper et al., 1989; Milich, 1988; Hohler et al., 1998]. Therefore the need of effective vaccine with fewer drawbacks is highly appreciated. This study aimed to use the polymerase protein of the HBV for multiple epitopes prediction that would act as vaccine candidates against the disease. The polymerase protein is considered as the only immunogene of Heptitis B virus that induced immune response[Mudawi, 2008; Bekele et al., 2015; Parkin and Cohen, 2001; Percus et al., 1993]. Multiple immunoinformatics approaches were exploited to investigate the efficacy of the chimeric vaccine construct.
One report by Depla et al (2008) described the design and synthesis of multi-epitopes vaccine against HBV in plasmid DNA construct and a recombinant MVA viral vector each contain a single gene encoding epitopes from cytotoxic and helper T lymphocytes. Their successful designed vaccine enhanced the immune responses of the host. Their results indicating the capability of multi-epitopes therapeutic vaccine in stimulating cellular immune responses and are essential for controlling and clearing HBV infection in in vitro and in HLA transgenic mice. Thus the coupled efforts of vaccination, interruption of transmission and the effective remedy are important factors to combat HBV [Chen 2009]
To generate the chimeric vaccine, short protein sequences act as linkers (spacers) were introduced between the B and T cells predicted epitopes to generate multi-epitopes peptides [Meza et al., 2017]. The linkers were reported to cause minimal junctional immunogenicity [Shey et al., 2019; Hasan et al., 2019; Ali et al., 2107;Pandey et al., 2018; Khatoon et al., 2017] and to ameliorate the bioactivity of the chimeric vaccine and to reach a high level of expression[Shey et al., 2019;Meza et al., 2017]. Moreover the chimeric vaccine was supported with an adjuvant in the N terminal of the vaccine. The adjuvants were previously reported as immunomodulator to ameliorate the activity of multiple vaccines [Mohan et al., 2013; Solanki and Tiwari, 2018]. It is noteworthy that bioinformatics and immunologic analysis tools provided that the chimeric vaccine should comprise MHCI and MHCII possessed epitopes in addition to linear and discontinuous B-cell epitopes [Ali et al., 2017]. Our chimeric vaccine was shown comprising linear and discontinuous B cell epitopes as well as MHC1 and MHCII epitopes. Also the vaccine construct was investigated for antigenicity and allergenicity. It demonstrated antigenicity in Vaxijen server and was shown to be nonallergic in Allertop server. This result indicated that the antigenic property of the vaccine without allergenicity further affirm its potentiality as a vaccine candidate. Furthermore the physical and chemical properties of the chimeric vaccine were analyzed.The analysis showed the protein was stable, contains aliphatic side chains, hydrophilic and demonstrated thermal stability. These features provided the suitability of the chimeric protein construct as good vaccine against the HBV.
Structural stability of protein secondary and tertiary structures are of crucial importance for efficient presentation of antigenic peptides on MHC for triggering strong immune reactions[Scheiblhofer et al., 2017]. Moreover fold stability directly impacted the existence of B-cell epitopes. At the same time protein destabilization leads to improper or unfoldingof the protein tertiary structure. This resulted in loss of conformational epitopes[Scheiblhofer et al., 2017]. Thus in this study the secondary and tertiary structure of the chimeric vaccine was investigated using multiple bioinformatics tools.The results showed that the secondary and tertiary structures were significantly important for generating a vaccine candidate. For instance the secondary structure of the chimeric protein contained alpha helices, extended strands, beta turns and random coils with no unambiguous or any other states. The 3D structure of the chimeric vaccine was ameliorated by the refined software and demonstrated desirable characteristics on Ramachandran plot predictions. The result indicated that most of the residues were in the favored areas with very minor residues in the outlier region, representing satisfactory model with good designed quality. One of the most cornerstones in designing vaccine is to analyze the solubility and the stability of the generated vaccine construct. It was previously reported that low solubility of the vaccine protein represents a drawback for production of large amounts of Hepatitis A virus proteins in BEVS vector[Silva et al., 2016]. In this study the solubility of the chimeric vaccine was measured compared to the solubility of E. coli proteins using protein sol server[Hebditch et al., 2017]. The solubility of the chimeric vaccine exceeds that of the E. coli proteins indicating the solubility of the chimeric protein. Moreover protein stability depends on disulfide bonds that considered as cornerstone forstructural folding and stability of a protein. Disulfide bonds changing the conformation of the protein based on the redox state of the environment and are critical for their folding. Disulfide bonds significantly reduced the number of conformations for a particular protein, resulting increased thermostability and decreased entropy[Berkmen, 2012; Zhang et al., 1994]. The chimeric protein in this study demonstrated five positions for disulfide bond formation depending on the chi3 residue screening, B-factor value and energy value. These five positions if mutated to cysteine would result in five disulfide bonds formation assisted in the stability of the vaccine construct. In addition to that, molecular dynamics analysis was performed to assess the complex stability of the chimeric vaccine. Previously a subset of atoms and covariance analysis has been analyzed for structural dynamics of proteins stability[Hasan et al., 2019; Aaltenetal., 1997]. Moreover previous studies correlated the stability of macromolecules with the fluctuation of atoms [Hasan et al., 2019; Caspar 1995; Clarage et al., 1995]. The molecular dynamics analysis of the chimeric vaccine in this study showed insignificant deformability of the vaccine residues strengthening the result of the chimeric protein stability.
Molecular docking between the chimeric vaccine protein and the TLR4 was performed to determine the favourable protein-protein interaction. The binding energies calculated from the interaction between our chimeric vaccine construct with TLR4 receptor further strengthen that the developed vaccine could potentially provoke protective immune response. It is of great significance is to express and validate the designed vaccine protein in suitable vector for immunoreactivity [Gori et al., 2013]. For the production of chimeric protein, E. coli expression systems are the most preferable choice [Rosano and Ceccarelli 2014; Chen, 2012]. Expression and translation of the vaccine construct in E. coli (strain K12) codon optimization was performed. The CAI was 0.9199, representing high proportion of most abundant codons. The GC- content was 51.58% representing high-level of expression of the vaccine protein in bacterial host.
Conclusion
Novel and effective vaccine against HBV is essentially required. The approach of reverse vaccinology was exploited in this study for designing potential multi epitopes vaccine from the polymerase protein of the HBV eliciting both B and T cells lymphocytes. The proposed epitopes were clonally expressed in the E. coli thus, providing the ability to work as putative vaccine candidates to combat the action of HBV.
Acknowledgments
Authors would like to thank the staff members of Department of Molecular Biology and Bioinformatics, College of Veterinary Medicine, University of Bahri/ Sudan for their cooperation and support.
Funding Sourse
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Competing Interest
The authors declare that they have no competing interests.
References
- FranckiI.B., FauquetC., Knudson D. Classification and Nomenclature of Viruses: Fifth Report of the International Committee on Taxonomy of Viruses. Virology Division of the International Union of Microbiological Societies.2012; Vol. 2: Springer Science & Business Media.
- Beasley R.P., Hwang L.Y., Lin C.C., Chien C.S. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan.Lancet. 1981; 21;2(8256):1129-33. doi: 10.1016/s0140-6736(81)90585-7. PMID: 6118576.
CrossRef - Shepard C.W., Simard E.P., Finelli L., Fiore A.E., Bell B.P. Hepatitis B virus infection: epidemiology and vaccination. Epidemiol Rev. 2006;28:112-25. doi: 10.1093/epirev/mxj009.
CrossRef - Ott J.J., Stevens G.A., Groeger J., Wiersma S.T. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAgseroprevalence and endemicity. Vaccine. 2012; 9;30(12):2212-9. doi: 10.1016/j
CrossRef - Yousif M., Mudawi H., Bakhiet S. et al. Molecular characterization of hepatitis B virus in liver disease patients and asymptomatic carriers of the virus in Sudan. BMC Infect Dis. 2013;13,328. https://doi.org/10.1186/1471-2334-13-328
CrossRef - Schattner A. Consequence or coincidence? The occurrence, pathogenesis and significance of autoimmune manifestations after viral vaccines.Vaccine. 2005; 10;23(30):3876-86. doi: 10.1016/j.vaccine.2005.03.005.
CrossRef - Rehermann B., Fowler P., Sidney J., Person J., Redeker A., Brown M., Moss B., Sette A., Chisari F.V. The cytotoxic T lymphocyte response to multiple hepatitis B virus polymerase epitopes during and after acute viral hepatitis. J Exp Med. 1995 Mar 1;181(3):1047-58. doi: 10.1084/jem.181.3.1047.
CrossRef - Mudawi H.M. Epidemiology of viral hepatitis in Sudan. ClinExpGastroenterol. 2008;1:9-13. doi: 10.2147/ceg.s3887.
CrossRef - Bekele Y., Amu S., Bobosha K., Lantto R., Nilsson A., Endale B., Gebre M., Aseffa A., RéthiB., Howe R., Chiodi F. Impaired phenotype and function of T follicular Helper cells in HIV-1-infected children receiving ART. Medicine.2015; 94.
CrossRef - Parkin J., Cohen B. An overview of the immune system.Lancet. 2001; 2;357(9270):1777-89. doi: 10.1016/S0140-6736(00)04904-7.
CrossRef - Percus J. K., PercusO. E., Perelson A. S. Predicting the size of the T-cell receptor and antibody combining region from consideration of efficient self-nonself discrimination.Proceedings of the National Academy of Sciences of the United States of America. 1993; 90(5), 1691–1695.
CrossRef - Enshell-Seijffers D., Denisov D., Groisman B., Smelyanski L., Meyuhas R., Gross G., Denisova G., Gershoni J.M. The mapping and reconstitution of a conformational discontinuous B-cell epitope of HIV-1. J Mol Biol. 2003; 14;334(1):87-101. doi: 10.1016/j.jmb.2003.09.002.
CrossRef - Potocnakova L., Bhide M., Pulzova L.B.An introduction to B-cell epitope mapping and in silico epitope prediction.Journal of immunology research.2016, Article ID 6760830, 11 pages http://dx.doi.org/10.1155/2016/6760830.
CrossRef - Frikha-Gargouri O., Gdoura R., Znazen A. et al. Evaluation of an in silico predicted specific and immunogenic antigen from the OmcB protein for the serodiagnosis of Chlamydia trachomatis BMC Microbiol. 2008; 8,217https://doi.org/10.1186/1471-2180-8-217
CrossRef - Hall T.A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. in Nucleic acids symposium series. [London]: Information Retrieval Ltd. 1999; c1979-c2000.
- Larsen J.E., Lund O., Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res.2006; 2:2.
CrossRef - Ponomarenko J.V., Bourne P.E. Antibody-protein interactions: benchmark datasets and prediction tools evaluation. BMC Struct Biol. 2007; 7:64.
CrossRef - Haste Andersen P., Nielsen M., Lund O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci.2006; 15:2558-2567.
CrossRef - Emini E.A., Hughes J.V., Perlow D.S., Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol.1985; 55:836-839.
CrossRef - Kolaskar A.S., Tongaonkar P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens.FEBS Lett.1990; 276:172-174.
CrossRef - JanewayC.A. Approaching the asymptote? Evolution and revolution in immunology.in Cold Spring Harbor symposia on quantitative biology. Cold Spring Harbor Laboratory Press. 1989
CrossRef - Kim Y., Ponomarenko J., Zhu Z., Tamang D., Wang P., et al. Immune epitope database analysis resource. Nucleic Acids Res. 2012; 438.
CrossRef - Nielsen M., Lundegaard C., Worning P., Lauemøller S.L., Lamberth K., et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci.2003; 12: 1007-1017.
CrossRef - Lundegaard C., Lamberth K., Harndahl M., Buus S., Lund O., et al. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008; 36: W509-W12.
CrossRef - Sidney J., AssarssonE.,Moore C., Ngo S., Pinilla C., et al. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 2008; 4: 2
CrossRef - Wang P., Sidney J., Dow C., Mothe B., Sette A.A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach.PLoSComput Biol.2008; 4: e1000048.
CrossRef - Dimitrov I., Bangov I., Flower D.R., Doytchinova I.A.AllerTOP v.2- a server for in silico prediction of allergens. J Mol. Model.2013; 20, 2278.
CrossRef - Gupta S., Kapoor P.,Chaudhary K., Gautam A., Kumar R. Open source drug discovery consortium, Raghava GP in silico approach for predicting toxicity of peptides and proteins. PLoS One.2013; 8 (9), e73957.
CrossRef - Shey R.A., Ghogomu S.M., Esoh K.K., Nebangwa N.D., Shintouo C.M., Nongley N.F., Asa B.F., Ngale F.N., Vanhamme L., Souopgui J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases.Sci Rep. 2019; 13; 9 (1):4409. doi: 10.1038/s41598-019-40833-x.
CrossRef - Hasan M., Ghosh P.P., Azim K.F., Mukta S., Abir R.A., Nahar J., Hasan Khan M.M. Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus. Microb Pathog.;130:19-37. doi: 10.1016/j.micpath.2019; 02.023. Epub 2019 Feb 26.
CrossRef - Nezafat N., Ghasemi Y., Javadi G., Khoshnoud M.J., Omidinia E. A novel multi-epitope peptide vaccine against cancer: an in silico approach. J. Theor. Biol. 2014; 349, 121–134.
CrossRef - Ali, PandeyR., KhatoonN., NarulaA., MishraA., PrajapatiV. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci Rep. 2017; 7, 9232.
CrossRef - Combet C., Blanchet C., GeourjonC. and Deléage G. NPS@ Network Protein Sequence Analysis TIBS. 2000; Vol. 25, No 3 [291]:147-150.
CrossRef - Kelley L.,Mezulis S., Yates C., et al.The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015; 10, 845–858. https://doi.org/10.1038/nprot.2015.053
CrossRef - Shin W.H., LeeG. R., Heo L., Lee H., and Seok C. Prediction of protein structure and interaction by GALAXY protein modeling programs, Bio Design. 2014; 2 (1), 1-11.
- KoJ., Park H., Heo L., and Seok C.Galaxy WEB server for protein structure prediction and refinement, Nucleic Acids Res. 2012; 40 (W1), W294-W297.
CrossRef - Lovell S.C., Davis I.W., Arendall W.B., Bakker P.I.W., Word J.M., Prisant M.G.,Richardson J.S. and Richardson D.C. Structure validation by Calpha geometry: phi, psiand C beta deviation, Protein Struct. Funct.Genet. 2002; 50, 437–450.
CrossRef - Al-Hakim M., Hasan R., Ali M.F., Rabbee J., Marufatuzzahan Z.F. In-silicocharacterization and homology modeling of catechol 1,2 dioxygenase involved inprocessing of catechol- an intermediate of aromatic compound degradationpathway, Glob. J. Sci. Front. Res. G Bio-Tech Genet. 2015; 15, 1–13.
- Wiederstein and SipplM.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res Jul; 35(Web Server issue).2007; W407–W410.
CrossRef - Ponomarenko J.V., Bui H., Li W., Fusseder N., Bourne P.E., Sette A., Peters B.ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics.2008; 9:514.
CrossRef - Hebditch M., Carballo-Amador M.A., Charonis S., Curtis R., Warwicker J.Protein-Sol: a web tool for predicting protein solubility from sequence.2017; 1;33(19):3098-3100. doi: 10.1093/bioinformatics/btx345.
CrossRef - Niwa T., Ying B.W., Saito K., Jin W., Takada S., Ueda T., Taguchi H.Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins, Proc. Natl. Acad. Sci. Unit. States Am. 2009; 106, 4201–4206.
CrossRef - Craig D.B.,Dombkowski A.A. Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinformatics .2013; 1; 14: 346. DOI: 1186/1471-2105-14-346 PMID: 24289175
CrossRef - Lopez-Blanco J.R., Aliaga J.I., Quintana-Orti E.S., Chacon P.iMODS: internal coordinates normal mode analysis server, Nucleic Acids Res. 2014; 42W271–W276.
CrossRef - AwanF.M., Obaid A., Ikram A., Janjua H.A. Mutation-structure function relationship based integrated strategy reveals the potential impact of deleterious missense mutations in autophagy related proteins on hepatocellular carcinoma (HCC): a comprehensive informatics approach, Int. J. Mol. Sci. 2017;18 (1) 139.
CrossRef - Prabhakar P.K., Srivastava A., Rao K.K., Balaji P.V.Monomerization alters the dynamics of the lid region in campylobacter jejuniCstII: an MD simulation study, J. Biomol. Struct.Dyn. 2016; 34 (4) 778–779.
CrossRef - Lopez-Blanco J.R., Garzon J.I., Chacon P.iMod, multipurpose normal mode analysis in internal coordinates, Bioinformatics. 2011; 27 (20) 2843–2850.
CrossRef - Vajda S., Yueh C., Beglov D., Bohnuud T., Mottarella S.E., Xia B., Hall D.R., Kozakov D. New additions to the ClusPro server motivated by CAPRI.Proteins: Structure, Function, and Bioinformatics. 2017; 85(3):435-444.
CrossRef - Kozakov D., Hall D.R., Xia B., Porter K.A., Padhorny D., Yueh C., Beglov D., Vajda S. The ClusPro web server for protein-protein docking.Nature Protocols.2017; Feb;12(2):255-278.
CrossRef - Morla S., Makhija A., Kumar S. Synonymous codon usage pattern in glycoprotein gene of rabies virus. Gene. 2016; 584, 1–6
CrossRef - Pandey R.K., Ojha R., Aathmanathan V.S., Krishnan M., Prajapati V.K.Immunoinformatics approaches to design a novel multi-epitope subunit vaccine against HIV infection, Vaccine. 2018; 36, 2262–2272
CrossRef - Hollinger F.B., Liang T.J. Hepatitis B virus. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, et al., editors. Fields Virology. Philadelphia, PA: Lippincott-Raven Publishers. 2001; pp. 2971–3036.
- Ward,Tang L.,Poonia B.,Kottilil S. Treatment of hepatitis B virus: an update. Future Microbiol. 2016; 11(12): 1581–1597.
CrossRef - Zheng J., Lin X., Wang X., Zheng L.,Lan S., Jin S., Ou Z., Wu J. In Silico Analysis of Epitope-Based Vaccine Candidates against Hepatitis B Virus Polymerase Protein.Viruses. 2017; 9, 112; doi:10.3390/v9050112
CrossRef - Lin C.L., Kao J.H. Review article: Novel therapies for hepatitis B virus cure—Advances and perspectives. Aliment.Pharmacol.Ther. 2016; 44, 213–222.
CrossRef - Jia H., Rai D., Zhan P., Chen X., Jiang X, Liu X. Recent advance of the hepatitis B virus inhibitors: A medicinal chemistry overview. Future Med. Chem. 2015; 7, 587–607.
CrossRef - Sitrin R.D., Wampler D.E., Ellis R.W. Survey of hepatitis B vaccines and their production processes. In: Ellis RW, editor. Hepatitis B vaccine in clinical practice. New York: Marcel Dekker. 1993; 83–101.
- McMahon B.J., Helminiak C., Wainwright R.B., Bulkow L., Trimble B.A,Wainright K., et al. Frequency of adverse reactions to hepatitis Bvaccine in 43618 persons. Am J Med. 1992; 92:254 – 6.
CrossRef - McAleer W.J., Buynak E.B., Maigetter R.Z., Wampler D.E., Miller W.J.,Hilleman M.R., et al. Human hepatitis B vaccine from recombinantyeast.Nature.1984; 307:178 – 81.
CrossRef - Craven D.E., Awdeh Z.L., Kunches L.M., Yunis E.J., Deinstag J.L., WernerB.G., et al. Nonresponsiveness to hepatitis B vaccine in health careworkers: results of revaccination and genetic typing. Ann Intern Med. 1986; 105:356 – 60.
CrossRef - Alper C.A., Kruskall M.S., Marcus-Bagley D., Craven D.E., Katz A.J.,Brink S.J., et al. Genetic prediction of non-responsiveness to hepatitis Bvaccine. N Engl J Med. 1989; 321:708 – 12.
CrossRef - Milich D.R.T and B cell recognition of hepatitis B viral antigens.Immunol Today. 1988; 9:380 – 91.
CrossRef - Hohler T., Meyer C.U., Notghi A., Stradmann-Bellinghausen B., Schneider P.M., Starke R., et al. Influence of major histocompatibility complex class II and T cell V beta repertoire on response toimmunization with HBsAg. Hum Immunol. 1998; 59:212 – 8
CrossRef - Depla E., Van der Aa A., Livingston B.D., Crimi C., Allosery K., De Brabandere V., Krakover J., Murthy S., Huang M., Power S., Babé L., Dahlberg C., McKinney D., Sette A., Southwood S., Philip R., Newman M.J., Meheus L. Rational design of a multiepitope vaccine encoding T-lymphocyte epitopes for treatment of chronic hepatitis B virus infections. J Virol. 2008;82(1):435-50. doi: 10.1128/JVI.01505-07.
CrossRef - Chen D.S. Hepatitis B vaccination: The key towards elimination and eradication of hepatitis B. J Hepatol. 2009;50(4):805-16. doi: 10.1016/j.jhep.2009.01.002.
CrossRef - Meza B., Ascencio F., Sierra-Beltrán A.P., Torres J.&Angulo C. A novel design of a multi-antigenic, multistage and multi-epitope vaccine against Helicobacter pylori: An in silico approach. Infection, Genetics and Evolution. 2017; 49, 309–317.
CrossRef - Khatoon N., Pandey R.K., Prajapati V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep. 2017; 7, 82-85.
CrossRef - Mohan T., Verma P., Rao D.N. Novel adjuvants & delivery vehicles for vaccines development: a road ahead, Indian J. Med. Res.2013;138 (5) 779.
- Solanki V., Tiwari V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacterbaumannii, Scientific report. 2018; 8 (1) 9044.
CrossRef - Scheiblhofer S., Laimer J., Machado Y., Weiss R., Thalhamer J. Influence of protein fold stability on immunogenicity and its implications for vaccine design. Expert Rev Vaccines. 2107; 16(5):479–89. doi:10.1080/14760584-1306441
CrossRef - Silva H.C. J., PestanaC.P., Galler R., Medeiros M.A. Solubility as a limiting factor for expression of hepatitis A virus proteins in insect cell-baculovirus system. MemInstOswaldo Cruz. 2016; 111(8):535‐ doi:10.1590/0074-02760160153
CrossRef - Berkmen M. Production of disulfide-bonded proteins in Escherichia coli. Protein Expression and Purification. 2012; 82(1), 240–251. doi:10.1016/j.pep.2011.10.009
CrossRef - Zhang T., Bertelsen E., Alber T. Entropic effects of disulphide bonds on protein stability, Nat. Struct. Biol.1994; (1) 434–438.
CrossRef - Aalten D.M.F., Groot B.L., Findlay J.B.C., Berendsen H.J.C., Amadei A.A comparison of techniques for calculating protein essential dynamics. J. Comput. Chem. 1997; 18 (2) 169–181.
CrossRef - Clarage J.B., Romo T., Andrews B.K., Pettitt B.M., Phillips G.N. A sampling problem in molecular dynamics simulations of macromolecules, Proc. Natl. Acad. Sci. U.S.A.1995; 92, 3288–3292.
CrossRef - Caspar D.L.D. Problems in simulating macromolecular movements, Structure.1995; 3, 327–329.
CrossRef - Gori A., LonghiR., Peri C. & Colombo G. Peptides for immunological purposes: design, strategies and applications. Amino Acids. 2013; 45, 257–268.
CrossRef - Chen R.Bacterial expression systems for recombinant protein production: E. coli and beyond. Biotechnol Adv.2012; 30, 1102–7.
CrossRef - Rosano G.L.,Ceccarelli E.A. Recombinant protein expression in Escherichia coli: advances and challenges. Frontiers in Microbiology.2014; 5, 172.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.



