
In – Silico Studies of Thiopyridine Compounds as Anti – Bacterial Agents Targeting Enoyl – Acyl Carrier Protein Reductase
 , Sandra Jose1, , Venkataraghavan Ragunathan2 and Sakthiselvan Punniavan1
, Sandra Jose1, , Venkataraghavan Ragunathan2 and Sakthiselvan Punniavan1 1Department of Bio-Engineering, School of Engineering, Vels Institute of Science Technology and Advanced Studies, Chennai-600117, Tamil Nadu, India
2Department of Chemical Engineering, Alagappa College of Technology, Anna University, Chennai-600025.
Corresponding Author E-mail: meenambiga.se@velsuniv.ac.in
DOI : http://dx.doi.org/10.13005/bbra/2962
Download this article as:
![]()
In the Fatty Acid Synthase II system, Enoyl-(acyl-carrier-protein) reductase (ENR) encoded by FabI genes is a limiting step enzyme and there is no homologue ENR found in invertebrates which makes it selective target for drug discovery. From Molecular dynamics simulations it was concluded that the solvated protein stabilized at 2.5 ns with larger mobility in the substrate - binding loop and the conformational flexibility of the molecule was revealed. To study the inhibitory effects of novel small molecules in the thiopyridine series, a 2D QSAR model was developed and evaluated for its efficiency. The R2 > 0.96 and Q2 = 0.978 depicted the predictive ability of the models which was determined using a test set of 3 compounds. The receptor-ligand interactions were studied and highest affinity was reported for GCT ID, 343129 (-9.09 Kcal/mol), 341772 (-8.90 Kcal/mol) and 268776 (-8.85 Kcal/mol). These compounds were analysed for their drug like properties and toxicity which projected acceptable blood brain barrier permeation and human intestinal absorption and reduced lipotoxicity. Thus the results suggest further synthesis of new thipyridine series of compounds and experimental testing against drug resistant Staphylococcal infections
KEYWORDS:ADMET analysis; ENR inhibition; Molecular dynamics; Molecular Docking; 2D-QSAR; Thiopyridine Series
Introduction
Our gut microbiota inhabits several kinds of Staphylococcus aureus strains, a principal causative agent associated with indwelling medical devices, which have achieved antibiotic resistance, most remarkable against methicillin and vancomycin 1, 2. There are unique genes that act as a vehicle for such species which guides them in niche adaptation even in unfavourable conditions and can even emerge as life-threatening [3]. Having conquered almost all the antibiotics that have been developed since the 1940s, S. aureus possesses unequalled machinery to trick and evade our adaptive immune response 4. The fatty acid synthesis is refined through one of the biosynthetic pathways, yet tantamount biosynthesis pathways for fatty acids, DNA, RNA, and protein has not been widely studied. The genes involved in the pathway are shown to be essential for normal functionality while considerable contrasts among the structures and the enzyme organisations catalyzing this pathway in vertebrates and microbes suggest that it is plausible to develop highly specific bacterial fatty acid synthesis inhibitors 5. Among several enzymes required for catalysing pathways, bacterial Enoyl-acyl carrier protein reductase (ENR), encoded by a highly conserved sequence among diverse bacterial species, FabI genes, is vital and an attractive target for antimicrobial drug discovery 6. These homologous FabI genes encode the entire ENR activity in gram-positive species such as S. aureus and gram-negative species including Escherichia coli and Haemophilus influenzae 7, 8. Time dependent interaction between biological molecules like protein-protein interactions, protein-ligand interactions is termed as biological activity.
To gain a notable insight into the intermolecular interactions in biomolecules, an atomic-level structural elucidation is exceedingly helpful. Such time-dependent microscopic behaviour of the bacterial enoyl-acyl carrier protein reductase is calculated by molecular dynamics (MD) simulation and MD simulations predict the movements of atoms present in a molecular system over time, based on their interatomic interactions 9.
Several in-vitro studies reveal the inhibitory potential of members of the thiopyridine chemical series against purified ENR which helps in the retardation of bacterial growth 10. Additionally, these compounds were also identified as cytotoxic agents which manifests induced apoptosis in human breast cancer cells 11. Hence, the thiopyridine series of compounds that inhibit Staphylococcal growth by inhibiting Enoyl acyl carrier protein Reductase (ENR) action in vitro were used to develop an in-silico model using QSAR studies, and docking studies were conducted on a set of test compounds 12.
Advancements in the concept of Quantitative Structure-Activity Relationship (QSAR) studies which is one of the conventional in-silico drug design approaches have made the drug development process rigorous and cost-effective. Our QSAR studies aimed at developing an excellent model for compounds that could inhibit Staphylococcal growth by inhibiting Enoyl acyl carrier protein Reductase (ENR), thus inhibiting the bacterial virulence.
In-silico studies on ENR inhibition with the thiopyridine series served as a good drug target for drug resistant Staphylococcal infections and the insights gained in this work could be used in further experimental studies.
Methods
Receptor and ligand preparation
The 3D structure of the protein FabI of Staphylococcus aureus was obtained from the Protein Data Bank (PDB ID: 3GR6) (https://www.rcsb.org/structure/3GR6) which is bound to NADP and triclosan [13, 14]. PRODRG (prodrg1.dyndns.org), a tool to generate the molecular topologies and unique molecular descriptors from coordinates of small molecules, operating on small molecules observed in the PDB collection of protein structures. Input obtained from previously existing coordinates or various two-dimensional formats is used to automatically generate coordinates and molecular topologies suitable for X-ray refinement of protein-ligand complexes, with which a 3D structure can be regenerated using energy minimization 15. The three-dimensional structures of the nine ENR inhibitors of the thiopyridine series selected in this study were generated by using PRODRG server and the output was saved as .pdb files and then converted to .smi file format using Corina.
Molecular Dynamics Simulation
GROMACS (GROningen Machine for Chemical Simulations) is a package for molecular dynamics simulation and energy minimization using which the topology files were created for the PDB structure of the molecule and were converted to GROMACS format 16. The solvent was added followed by initial energy minimization and equilibration run. The ngmx program is a special visualization program of GROMACS which reads a trajectory file and an index file and plots a 3D structure of our molecule on our standard X window screen. Grace, a WYSIWYG 2D graph plotting tool for UNIX-like operating systems, use the X window system and Motif for its GUI for creating publication-quality output 17.
Ligand Selection and descriptor calculation
Potential inhibitory properties-based literature review suggested a dataset of 9 compounds of thiopyridine series whose preliminary structure-activity relationships, potency on purified ENR, and activity on bacterial cells comprehend the 9 ligands as effective fatty acid biosynthesis inhibitors suitable for further antibacterial development 18, 19. The structure, molecular formula, and their biological activity (log IC50values) of the training and testing set compounds are tabulated in Table 1.
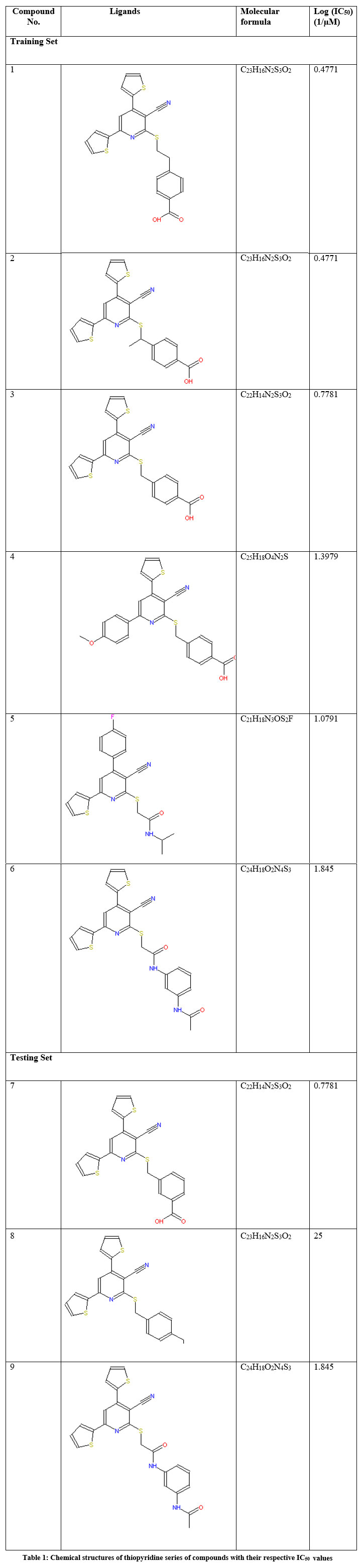 |
Table 1: Chemical structures of thiopyridine series of compounds with their respective IC50 values. |
E-DRAGON (http://www.vcclab.org/lab/edragon/), an electronic software that helps to calculate the molecular descriptors which can be used to evaluate molecular structure-activity or structure-property relationships, similarity analysis and also for high throughput screening of molecule databases 20, 21. Dragon requires 3D optimized structures with hydrogen. The .smi files of all the compounds were given as input to the E-Dragon software to generate various theoretical molecular descriptors. The descriptors calculated were tabulated in an excel file format to be given as input to the Simca P+ software for QSAR model development.
QSAR Analysis and Model Development
QSAR studies were performed using Simca P+ software. The Inhibition value (IC50) was taken as the dependent variable and the various descriptors generated were taken as independent variables. A classical QSAR model was initially generated by multiple linear regression methods taking only QSAR properties as independent variables 22. Principal Component Analysis was performed on a whole set of descriptors generated to determine the correlation between the variables and compress the data to generate a new set of variables called Principal Components 23. These optimum numbers of principal components which would provide smallest standard error of prediction were determined by the leave-one-out cross-validation procedure. The Principal components extracted were further subjected to regression by the Partial least squares method and the R2 (The variation in data explained by the model) and Q2 (Predictive power of the model) values were determined The R2 and Q2 value of the final model were determined from the output. The model is accepted to be a good model if Q2 is greater than 0.5 and extremely good if Q2 is greater than 0.9. The model can be concluded to be a valid one if the R2Y intercept is less than 0.4 and the Q2 intercept is less than 0.05 and finally the training set was used to cross-validate the model obtained.
Molecular Docking
Computational analysis of protein ligand interactions was performed using Autodock 4.2.6 (http://autodock.scripps.edu/) 24. The 3D structure of FabI of Staphylococcus aureus was obtained from the Protein Data Bank (PDB ID: 3GR6). The macromolecule and the ligands were prepared by adding polar hydrogen and Kollman charges and saved 25. An extended PDB format, termed PDBQT, is used for coordinate files, which includes atomic partial charges and atom types. PDBQT files also include information on the torsional degrees of freedom. A grid box was set for blind docking to compute the atoms inside each grid. The binding energy was obtained for each ligand and the contact analysis of the docked complexes was done using WHATIF 25. The lower binding energy reveals higher stability of the bound conformation.
ADME-Toxicity drug-likeness Study
The Adsorption, Distribution, Metabolism, Excretion, and Toxicology properties filter the compounds based on their drug-likeness and play a significant role in the drug design process. 60% of all drugs in the clinical phases fail due to the neglected physicochemical characteristics of the molecules. The updated version of admetSAR, admetSAR 2.0 (http://lmmd.ecust.edu.cn/admetsar2/) is used to predict advanced toxicity traits such as human oral bioavailability, carcinogenicity, hepatotoxicity, fish aquatic toxicity, and biodegradation 26. The top-scoring molecules with the best docking scores are selected for ADMET analysis and the results are tabulated.
Results
The Molecular Dynamics Simulation of Enoyl Acyl carrier protein Reductase
Molecular dynamics simulation of Fab I protein was performed using GROMACS 4.0 and the average RMSD was calculated and the graph was drawn using GRACE. The RMSD value steadily increased, stabilized around 2.5 ns, and remained so till the end of 5 ns. The RMSD plot is shown in Figure 1. The Root Mean Square Fluctuation Graph shown in Figure 2 was drawn to analyze the ligand-binding loop and the other flexible regions of the enzyme. The maximum fluctuations were observed in the 200 to 220 residues. Generally, the α-carbon of secondary structures of the protein fluctuate more compared the loop and turn regions.
 |
Figure 1: RMSD plot of Molecular Dynamics Simulation. |
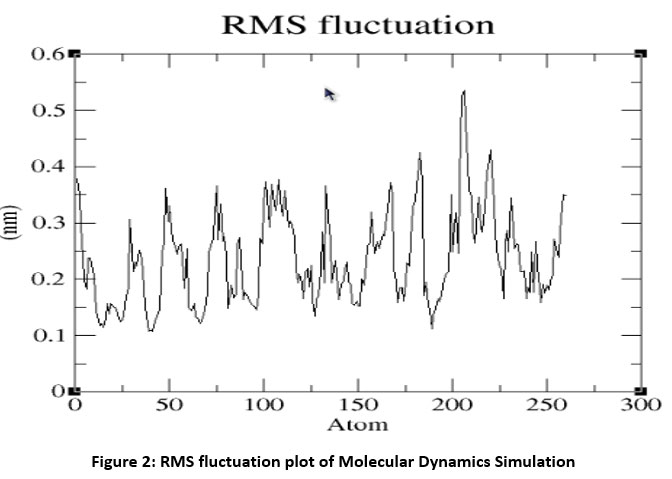 |
Figure 2: RMS fluctuation plot of Molecular Dynamics Simulation |
QSAR Analysis
The biological activity (log (IC50) values) along with the descriptors obtained from E-dragon software, were imported into the Simca P+ software and the descriptors that give the best model was identified. The descriptors thus selected for QSAR model development and their values are tabulated in Table 2.
Table 2: Descriptors selected for QSAR analysis and their values
| S. No | Molecular Descriptor | R2X | R2Y | Q2 |
| 1 | Constitutional descriptor | 0.56 | 0.707 | 0.397 |
| 2 | Molecular descriptor | 0.916 | 0.989 | 0.901 |
| 3 | Information indices | 0.806 | 0.911 | 0.679 |
| 4 | Edge adjacency indices | 0.985 | 0.934 | 0.826 |
| 5 | WHIM descriptors | 0.755 | 0.966 | 0.659 |
| 6 | Topological descriptor | 0.842 | 0.944 | 0.772 |
| 7 | RDF descriptors | 0.561 | 0.994 | 0.671 |
| 8 | Geometrical descriptor | 0.5 | 0.933 | 0.599 |
| 9 | Functional group counts | 0.533 | 0.73 | 0.455 |
| 10 | Eigenvalue based indices | 0.873 | 0.875 | 0.615 |
The correlation was established between these descriptors and the biological activity of the training set using multiple linear regressions. The regression analysis resulted in various R2values (variation in IC50 value) and Q2 values (model predictivity). From the analysis, it was concluded that the edge adjacency indices descriptors, molecular properties descriptors, and topological descriptors have some contribution towards model predictivity as their R2 value and Q2 values were above 0.9 and were correlated with biological activity. The least correlated variables from the model were eliminated the number of variables was minimized to 5, MLogP, Espm01d, EEig10x, S2K, and PHI. The model values were upright with R2 > 0.96 and Q2 = 0.978. The score plot and variable importance plot of the model are given in Figure 3 and Figure 4 respectively.
 |
Figure 3: Score plot of the model for the training set. |
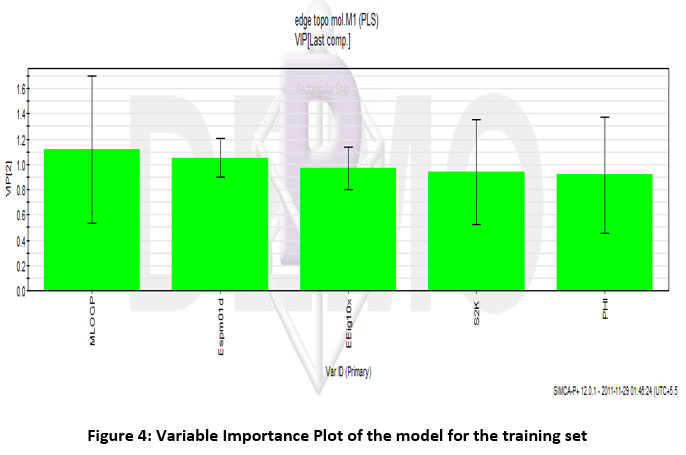 |
Figure 4: Variable Importance Plot of the model for the training set. |
The predicted log (IC50) values obtained from the study are more or less equal to the observed values and hence the test set compounds can be used for prediction of biological activity of similar compounds to develop experimental leads. Analysis comparative of observed and predicted log (IC50) values for the test set are given in table 3.
Table 3: Observed Vs Predicted log (IC50) values for the test set
| Compound No. | Predicted log (IC50) values | Observed log (IC50) values |
| 7 | 0.6978 | 0.7181 |
| 8 | 1.0710 | 1.3979 |
| 9 | 1.02709 | 1.0791 |
Figure 5 is the model generated using the PLS method which showed an R2 intercept of 0.325 and a Q2 intercept of -0.48 which are lesser than 0.4 and 0.05 respectively which showcase a valid model to predict the biological activity of thiopyridine series of compounds for ENR activity inhibition.
 |
Figure 5: Validation plot from the response permutation test of the model generated by PLS method. |
Prediction of Biological Activity for compounds with similar structures
Structures of compounds similar to thiopyridine series were taken from the Zinc database and PubChem and their descriptors such as MLogP, Espm01d, EEig10x, S2K, and PHI were calculated using E-Dragon software. The QSAR model obtained was used to predict the IC50 values for these compounds using Simca P+ software. Structurally related compounds were given the same log (IC50) values and by trial and error method, the log (IC50) values were fed until we get the model values of R2 > 0.96 and Q2 = 0.978. The chemical structure and the predicted log (IC50) values are tabulated in Table 4. These predicted biological activity values are similar to the values of test set compounds proving the validity of the model to study similar structures that can be used as experimental leads for developing new compounds for ENR inhibition.
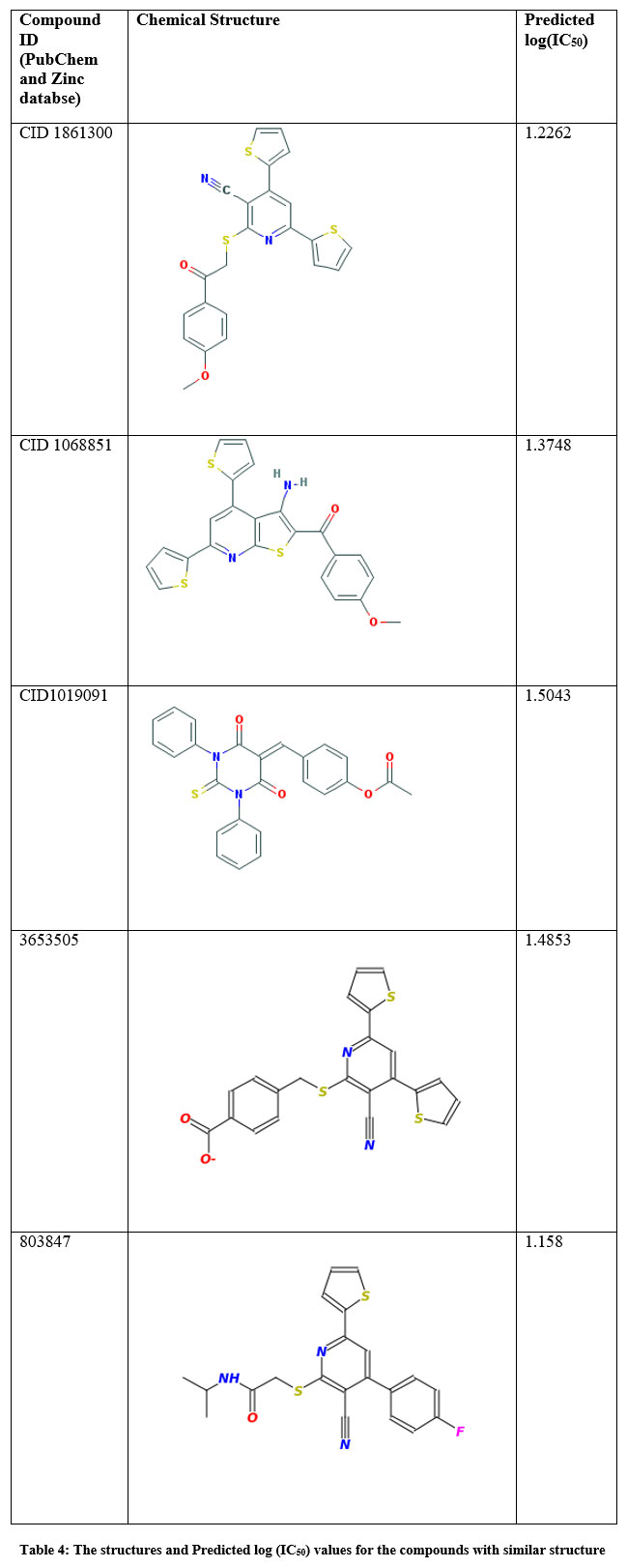 |
Table 4: The structures and Predicted log (IC50) values for the compounds with similar structure. |
Molecular Docking Results
In order to analyze the interatomic and intermolecular interactions of the ligands, the training set compounds were docked to the structure of ENR of Staphylococcus aureus. The corresponding docked score, the ligands’ hydrophobic and hydrogen contacts are shown in Table 5. The molecular docking provides various insights to study the target-inhibitor interactions and possible mechanism of the same.
Table 5: Docked score and the Interatomic contacts of the training set compounds
| Compound ID | Docking score (kcal/mol) | Hydrogen contacts | Hydrophobic contacts |
| 1 | -9.09 | Ala 95, Ser 197
|
Gly 13, Ala 15, Ile 20, Ser 93, Ile 94, Ala 95, Thr 145, Tyr 147, Tyr 157, Met 160, Lys 164, Thr 195, Leu196, Ser 197, Val 201. |
| 2 | -8.65 | Ala 95, Ser 197 | Gly 13, Ser 19, Ile 20, Ala 21, Ser 93, Ile 94, Ala 95, Thr 145, Thr 146, Tyr 147, Tyr 157, Lys 164, Gly 191, Pro 192, Ile 193, Thr 195, Ala 198, Val 201, Phe 204. |
| 3 | -8.32 | Ala 95, Thr 195 | Ala 15, Ser 19, Ile 20, Ala 21, Arg 40, Ser 93, Ile 94, Phe 96, Thr 145, Lys 164, Ile 193, Leu 196, Ser 197, Ala 198. |
| 4 | -8.90 | Ser 197 | Gly 13, Ala 15, Ile 20, Ser 93, Ile 94, Ala 95, Thr 145, Tyr 147, Tyr 157, Met 160, Thr 195, Leu 196, Ser 197, Val 201. |
| 5 | -8.85 | Ala 95, Lys 164, Leu 196 | Gly 13, Ala 15, Asn 16, Arg 18, Ser 19, Ile 20, Arg 40, Ser 93, Ile 94, Thr 145, Thr 146, Tyr 147, Ala 190, Thr 195. |
| 6 | -8.23 | Ala 95, Lys 164 | Gly 13, Arg 18, Ser 19, Ile 20, Arg 40, Ser 93, Ile 94, Phe 96, Thr 145, Thr 146, Tyr 147, Ala 190, Gly 191, Pro 192, Thr 195, Leu 196, Ser 197, Phe 204. |
Docking studies indicated that ligands bind to the catalytic site of FabI with considerable binding energy while all the ligands were bound in the same hydrophobic pocket. Compound 1 showed good binding energy of -9.09 kcal/mol with two conventional hydrogen bond contacts with Ala 95 and Ser 197 within 3Å followed by compound 4 (-8.90 kcal/mol), and compound 5 (-8.85 kcal/mol) against ENR enzyme. The residues which form hydrogen bonds with the ligands were Ala 95, Lys 164, Ile 193, Thr 195, Leu 196 and Ser 197. The hydrophobic contacts were Gly 13, Ala 15, Ser 19, Ile 20, Ala 21, Arg 40, Ser 93, Ile 94, Phe 96, Thr 145, Thr 146, Tyr 157, Met 160, Ala 190, Gly 191, Pro192, Ala 198, Val 201, and Phe 204. Figure 6 shows the three-dimensional plot for FabI-Thiopyridine interactions.
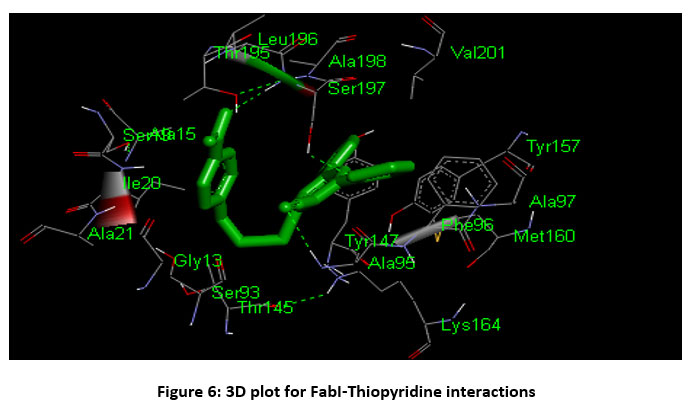 |
Figure 6: 3D plot for FabI-Thiopyridine interactions. |
ADME-TOX study of the top three compounds
The top-scoring compounds (1,4 and 5) were analysed for their ADMET properties. ADMET prediction inferred that the compounds follow Lipinski’s rule and fell in the applicability domain 27. The plasma protein binding capability of the compounds is less than 90% which indicates that these compounds have good bioavailability at the target organs. 1-9% cases of bacterial meningitis and 14-77% mortality are caused by S. aureus which can potentially lead to heart valve infection. Cases of Staphylococcal meningitis can be prevented drugs possessing good Blood-Brain barrier permeation. In addition to blood-brain barrier permeation these compounds fairly follow Lipinski’s rule (<5 Hydrogen Bond donors, <10 Hydrogen Bond Acceptors, Molecular mass <500Da and Log P<5) 28. The drug-likeness profile of the compounds is well depicted in table 6.
Table 6: ADMET Predicted Profiles with their drug like properties
| ADMET properties | Compound 1 | Compound 4 | Compound 5 |
| Molecular Weight | 447.59 | 441.49 | 411.53 |
| AlogP | 5.11 | 4.54 7 | 5.1 |
| H-Bond Acceptor | 7 | 7 | 5 |
| H-Bond Donor | 0 | 0 | 1 |
| Rotatable Bonds | 7 | 7 | 6 |
| Applicability Domain | In domain | In domain | In domain |
| Water solubility | -3.566 | -3.718 | -3.625 |
| Plasma protein binding | 0.851 | 0.861 | 0.9 |
In-silico toxicology tests like, Ames mutagenesis and carcinogenicity were tested negative for all the three compounds and they showed blood brain barrier permeation. Acceptable values of water solubility were reported which led to good predicted human intestinal absorption. Tetrahymena pyriformis (pIGC50) toxicity was reported as 1.017, 0.913 and 0.137 µg/L for compounds 1, 4 and 5 respectively. Several other parameters like Androgen receptor binding, Caco-2 cell permeability, Estrogen receptor binding, Glucocorticoid receptor binding, Hepatotoxicity and substrate inhibition of Cytochromes P450 (CYPs) superfamily are represented in table 7.
Table 7: Toxicity Studies of the top three compounds with highest binding affinities.
| Toxicity Test | Compound 1 | Compound 4 | Compound 5 |
| Ames mutagenesis | – | – | – |
| Acute Oral Toxicity (kg/mol) | 2.647 | 2.637 | 2.629 |
| Androgen receptor binding | + | + | + |
| Aromatase binding | + | + | + |
| Avian toxicity | – | – | – |
| Blood Brain Barrier | + | + | + |
| BRCP inhibitior | – | – | – |
| Biodegradation | – | – | – |
| BSEP inhibitior | + | + | + |
| Caco-2 | – | – | – |
| Carcinogenicity (binary) | – | – | – |
| crustacea aquatic toxicity | + | + | – |
| CYP1A2 inhibition | + | + | + |
| CYP2C19 inhibition | – | + | + |
| CYP2C9 inhibition | + | + | + |
| CYP2C9 substrate | – | – | – |
| CYP2D6 inhibition | – | – | – |
| CYP2D6 substrate | – | – | – |
| CYP3A4 inhibition | – | + | – |
| CYP3A4 substrate | + | + | + |
| CYP inhibitory promiscuity | + | + | + |
| Eye corrosion | – | – | – |
| Eye irritation | – | – | – |
| Estrogen receptor binding | + | + | + |
| Fish aquatic toxicity | + | + | + |
| Glucocorticoid receptor binding | + | + | + |
| Honey bee toxicity | – | – | – |
| Hepatotoxicity | + | + | + |
| Human either-a-go-go inhibition | + | + | + |
| Human Intestinal Absorption | + | + | + |
| Human oral bioavailability | – | + | – |
| MATE1 inhibitior | – | – | – |
| Micronuclear | + | + | + |
| OATP1B1 inhibitior | + | + | + |
| OATP1B3 inhibitior | + | + | + |
| OATP2B1 inhibitior | – | – | – |
| OCT1 inhibitior | – | – | – |
| OCT2 inhibitior | – | – | – |
| P-glycoprotein inhibitior | + | + | + |
| P-glycoprotein substrate | – | – | – |
| PPAR gamma | + | + | + |
| Subcellular localization | Mitochondria | Mitochondria | Mitochondria |
| Tetrahymena pyriformis (pIGC50) (ug/L) | 1.017 | 0.913 | 0.137 |
| Thyroid receptor binding | + | + | + |
| UGT catalyzed | – | – | – |
Discussion
Methicillin-resistant Staphylococcus aureus (MRSA) infection remains a challenging public health issue that causes multiple clinical emergencies like sepsis, pneumonia, meningitis, endocarditis, and toxic shock syndrome 29. First-line drugs used for MRSA infections include Vancomycin but in recent times, S. aureus isolates have evolved with complete resistance to the drug 30, 31. Time-dependent individual atomic motions of a protein molecule can help us study and understand the properties of a protein system and how the biomolecule functions [32]. Molecular dynamics simulations of the FabI of Staphylococcus aureus (PDB ID : 3GR6) obtained from PDB provided the explanations of the actual performance of molecules in real-time dynamics and the energy terms parameterized to fit quantum-mechanical estimations and analytical data. Molecular Dynamics Simulation Studies revealed that the ENR enzyme stabilizes at 2.5 ns with larger mobility in the substrate-binding loop. Also, there are mobile regions in close proximity to the substrate-binding loop which perhaps help in providing substrate access to the active site. This conformational flexibility has provided valuable insights into the domain movements and the binding modes of the inhibitors.
2D-QSAR models are used to predict the biological, chemical, and physical properties of molecules from their well-studied chemical structures. Ligand-based QSAR approaches coupled with site targeted receptor-ligand docking aids in obtaining highly accurate results for hypothetical receptor binding for undocked entries 33. Upon completion of the molecular dynamics simulations, a 2D-QSAR model was developed for the prediction of the thiopyridine series for Enoyl Acyl carrier protein reductase inhibitory activity. Thiopyridine, an antitumor agent of which the derivatives were also found to be highly effective in vitro against both actively proliferating and inactive non-culturable M. tuberculosis[34]. Enoyl-ACP reductase, currently an attractive drug target for tuberculosis treatment is involved in fatty acid synthesis hence playing a significant role in completing fatty acid biosynthesis cycles 35. In a similar study on ENR enzyme, the CoMFA model demonstrated Q2 = 0.631, R2 = 0.755 and Topomer CoMFA model exhibited Q2 = 0.644, R2 = 0.865 36. The current work aimed at discovering the thiopyridine compounds from Staphylococcus aureus as an effective Enoyl-ACP reductase inhibitor through molecular docking studies.
Molecular docking is a very appropriate tool to understand receptor-ligand interactions with high accuracy 38. The main objective is to computationally optimize the conformations to obtain the overall minimized free energy system. Docking studies indicated that the novel small molecules bind to the catalytic site of FabI in the same hydrophobic pocket with appreciable binding energy. The ligands extended hydrogen bonds with Ala 95, Lys 164, Ile 193, Thr 195, Leu 196, and Ser 197, and the hydrophobic contacts were Gly 13, Ala 15, Ser 19, Ile 20, Ala 21, Arg 40, Ser 93, Ile 94, Phe 96, Thr 145, Thr 146, Tyr 157, Met 160, Ala 190, Gly 191, Pro192, Ala 198, Val 201 and Phe 204. The importance of the residues that showed the highest mobility is predicted from the interactions of triclosan and NADP with the SaFabI enzyme 37. The largest mobility in the graph represents the substrate binding loop of the enzyme. Residues 198-205 represent the more ordered flipping loop of Staphylococcus aureus FabI enzyme. Ser 197, Val 201, and Phe 204 play a pivotal role in this substrate-binding loop. Residues 40-54 represents the region responsible for catalytic activity as the residues Arg 40 and Lys 41 lie very close to the phosphate moiety of the NADP cofactor. Residues 65-80 represent a flexible region as the residues Asp 66 and Val 67 form hydrophobic interactions with the ligand. Residues 94-120 form hydrophobic pockets important for nicotinamide binding formed by the hydrophobic residues such as Ile 94, Ala 95, and Ala 97. In addition to these residues obtained from literature, residues 171-188 and 220-225 show mobility. These are in close proximity to the substrate-binding loop. The movements in these regions might perhaps help in providing substrate access to the substrate – binding loop. Triclosan, an effective inhibitor of ENR, interacts with Ala 95, Tyr 157, Met 160, Ser 197, Ala 198, Val 201, and Phe 204 residues of FabI. The top three binding ligands were studied for their ADMET properties using admetSAR. No mutagenic or carcinogenic properties have been reported for the compounds. These compounds reported acceptable blood-brain barrier permeation and reduced lipotoxicity. Notably identified as subcellular localization in mitochondria, the compounds also gave positive PPAR-gamma activity which activates the PON1 gene which aids in reducing atherosclerosis [39]. Often, lower PPAR-gamma activity leads to lipotoxicity. All the three compounds possess excellent drug-like nature and fall in the applicability domain. These findings in correlation with the interactions of our ligand indicate that they bind to the suitable pocket and can be effectively explored for their potential for ENR inhibition. Inhibitor compounds with new scaffolds can be designed and optimized using the developed 2D-QSAR model.
Conclusion
Thiopyridine series of compounds that inhibit Staphylococcal growth by inhibiting Enoyl acyl carrier protein Reductase (ENR) action in vitro were used to develop an in-silico model using Quantitative Structure Activity Relationship Studies. Molecular dynamics simulations provided insights into conformational flexibility and functional specificity of the protein system including the domain movements and the binding modes of the inhibitors. Our QSAR studies on biological activity resulted in very good model predictivity and can be used to predict the activity. Compounds with predicted biological activity can be used as experimental leads for the discovery of new ENR inhibitors. Molecular Docking Analysis showed that the inhibitors bind to the catalytic site of ENR and the interactions were similar to the effective inhibitor Triclosan interactions with the enzyme. Thus, the in-silico model developed and effective binding of thiopyridine series inhibitors to ENR with minimal toxic effects reported from ADMET studies reveals that these compounds can be effectively explored as potential inhibitors in Staphylococcal infections. In-vitro ENR inhibition with thiopyridine series should be explored for drug-resistant Staphylococcal infections. We hope the insights gained in this work can be used in experimental studies.
Acknowledgement
The authors sincerely thank Vels Institute of Science Technology and Advanced Studies management for helping us towards the successful completion of our research work.
Conflicts of Interest
No conflicts of interest declared.
Funding Sources
There are no funding source.
References
- Sakoulas G, Eliopoulos GM, Moellering RC, Wennersten C, Venkataraman L, Novick RP, Gold HS. Accessory gene regulator (agr) locus in geographically diverse Staphylococcus aureus isolates with reduced susceptibility to vancomycin. Agents Chemother. 2002; 46(5):1492-502.
CrossRef - Vuong C, Kocianova S, Yao Y, Carmody AB, Otto M. Increased colonization of indwelling medical devices by quorum-sensing mutants of Staphylococcus epidermidis in vivo. Infect. Dis. 2004; 190(8):1498-505.
CrossRef - Lindsay JA. Genomic variation and evolution of Staphylococcus aureus. J. Med. Microbiol. 2010; 300(2-3): 98-103.
CrossRef - Kane TL, Carothers KE, Lee SW. Virulence factor targeting of the bacterial pathogen Staphylococcus aureus for vaccine and therapeutics. Drug Targets. 2018; 19(2): 111-27.
CrossRef - Parsons JB, Rock CO. Is bacterial fatty acid synthesis a valid target for antibacterial drug discovery?. Opin. Microbiol. 201; 14(5): 544-9.
CrossRef - Massengo-Tiassé RP, Cronan JE. Diversity in enoyl-acyl carrier protein reductases. Mol. Life Sci. 2009; 66(9): 1507-17.
CrossRef - Kini SG, Bhat A, Pan Z, Dayan FE. Synthesis and antitubercular activity of heterocycle substituted diphenyl ether derivatives. Enzyme Inhib. Med. Chem. 2010; 25(5): 730-6.
CrossRef - Marcinkeviciene J, Jiang W, Kopcho LM, Locke G, Luo Y, Copeland RA. Enoyl-ACP reductase (FabI) of Haemophilus influenzae: steady-state kinetic mechanism and inhibition by triclosan and hexachlorophene. Biochem. Biophys. 2001; 390(1):101-8.
CrossRef - Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron. 2018; 99(6): 1129-43.
CrossRef - Lu X, Huang K, You Q. Enoyl acyl carrier protein reductase inhibitors: a patent review (2006–2010). Expert Opin. Ther. Pat. 2011; 21(7): 1007-22.
CrossRef - Mandal S, Bérubé G, Asselin É, Mohammad I, Richardson VJ, Gupta A, Pramanik SK, Williams AL, Mandal SK. A novel series of potent cytotoxic agents targeting G2/M phase of the cell cycle and demonstrating cell killing by apoptosis in human breast cancer cells. Med. Chem. Lett. 2007; 17(17): 4955-60.
CrossRef - Ling LL, Xian J, Ali S, Geng B, Fan J, Mills DM, Arvanites AC, Orgueira H, Ashwell MA, Carmel G, Xiang Y. Identification and characterization of inhibitors of bacterial enoyl-acyl carrier protein reductase. Antimicrobial agents and chemotherapy. 2004 May;48(5):1541.
CrossRef - Rose PW, Prlić A, Altunkaya A, Bi C, Bradley AR, Christie CH, Costanzo LD, Duarte JM, Dutta S, Feng Z, Green RK. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2016; 45: D271-D281.
- Burley SK, Berman HM, Bhikadiya C, Bi C, Chen L, Di Costanzo L, Christie C, Dalenberg K, Duarte JM, Dutta S, Feng Z. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019; 47(D1): D464-74.
CrossRef - Schüttelkopf AW, Van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004; 60(8):1355-63.
CrossRef - Van der Spoel D, Lindahl E, Hess B, van Buuren AR, Apol E, Meulenhoff PJ, Tieleman DP, Sijbers AL, Feenstra KA, van Drunen R, Berendsen HJ. Gromacs: Groningen machine for chemical simulations.
- Turner P, Stambulchik E, Unix-Like A. Grace (plotting tool).
CrossRef - Pamudi BF, AZIZAHWATI AY. In-silico screening against antimalarial target plasmodium falciparum enoyl-acyl carrier protein reductase. Asian J. Pharm. Clin. Res. 2017; 10: 127-9.
- Lu XY, You QD, Chen YD. Recent progress in the identification and development of InhA direct inhibitors of Mycobacterium tuberculosis. Mini-Rev. Med. Chem. 2010; 10(3): 182-93.
CrossRef - Mauri A, Consonni V, Pavan M, Todeschini R. Dragon software: An easy approach to molecular descriptor calculations. MATCH. 2006; 56(2): 237-48.
- Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, Palyulin VA, Radchenko EV, Zefirov NS, Makarenko AS, Tanchuk VY. Virtual computational chemistry laboratory–design and description. Comput.-Aided Mol. Des. 2005; 19(6):453-63.
CrossRef - Afantitis A, Melagraki G, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O. A novel simple QSAR model for the prediction of anti-HIV activity using multiple linear regression analysis. Divers. 2006; 10(3): 405-14.
CrossRef - Hemmateenejad B, Miri R, Elyasi M. A segmented principal component analysis—regression approach to QSAR study of peptides. Theor. Biol. 2012 Jul 21;305:37-44.
CrossRef - Hou X, Du J, Zhang J, Du L, Fang H, Li M. How to improve docking accuracy of AutoDock4. 2: a case study using different electrostatic potentials. Chem. Inf. Model. 2013; 53(1): 188-200.
CrossRef - Vriend G. WHAT IF: a molecular modeling and drug design program. Mol. Graphics. 1990;8(1):52-6.
CrossRef - Yang H, Lou C, Sun L, Li J, Cai Y, Wang Z, Li W, Liu G, Tang Y. admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. 2019; 35(6):1067-9.
CrossRef - Lipinski CA. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technol. 2004; 1(4): 337-41.
CrossRef - Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Drug Delivery Rev. 1997; 23(1-3): 3-25.
CrossRef - Turner NA, Sharma-Kuinkel BK, Maskarinec SA, Eichenberger EM, Shah PP, Carugati M, Holland TL, Fowler VG. Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat. Rev. Microbiol. 2019; 17(4):203-18.
CrossRef - Thati V, Shivannavar CT, Gaddad SM. Vancomycin resistance among methicillin resistant Staphylococcus aureus isolates from intensive care units of tertiary care hospitals in Hyderabad. Indian J. Med. Res. 2011; 134(5):704-708.
CrossRef - Tarai B, Das P, Kumar D. Recurrent challenges for clinicians: emergence of methicillin-resistant Staphylococcus aureus, vancomycin resistance, and current treatment options. Lab. Physicians. 2013; 5(2):71-78.
CrossRef - Karplus M, Kuriyan J. Molecular dynamics and protein function. Natl. Acad. Sci. U. S. A. 2005; 102(19): 6679-85.
CrossRef - Roy K, Narayan Das R. A review on principles, theory and practices of 2D-QSAR. Drug Metab. 2014; 15(4): 346-79.
CrossRef - Salina EG, Ryabova O, Vocat A, Nikonenko B, Cole ST, Makarov V. New 1-hydroxy-2-thiopyridine derivatives active against both replicating and dormant Mycobacterium tuberculosis. J. Infect. Chemother. 2017; 23(11):794-7.
CrossRef - Marrakchi H, Zhang YM, Rock CO. Mechanistic diversity and regulation of Type II fatty acid synthesis. Soc. Trans.2002; 30(6): 1050–1055.
CrossRef - Joshi SD, Dixit SR, More UA, Kumar D, Aminabhavi TM, Kulkarni VH. 3D-QSAR studies of quinoline Schiff bases as enoyl acyl carrier protein reductase inhibitors. Reports Med. Chem. 2014; 4: 59-75.
CrossRef - Priyadarshi A, Kim EE, Hwang KY. Structural insights into Staphylococcus aureus enoyl‐ACP reductase (FabI), in complex with NADP and triclosan. Proteins: Struct., Funct., Bioinf. 2010; 78(2):480-6.
CrossRef - Meenambiga, S.S., Venkataraghavan, R. and Biswal, R.A., 2018. In silico analysis of plant phytochemicals against secreted aspartic proteinase enzyme of Candida albicans.J Appl Pharm Sci, 8(11), pp.140-50.
CrossRef - Stancu, C.S., Sanda, G.M., Deleanu, M. and Sima, A.V., 2014. Probiotics determine hypolipidemic and antioxidant effects in hyperlipidemic hamsters. Molecular nutrition & food research, 58(3), 559-568.
CrossRef
Accepted on: 16-11-2021
Second Review by: Dr. Naman Wadhwa
Final Approval by: Dr. Jahwarhar Izuan Bin Abdul Rashid








![]()
![]()
