

How to Cite | Publication History | PlumX Article Matrix
Harsh Kashyap1 , Vanshika Makol2 and Manisha Khatri2*
, Vanshika Makol2 and Manisha Khatri2*
1Amity Institute of Biotechnology, Amity University, Noida, Uttarpradesh India.
2Department of Biomedical Science, Shaheed Rajguru College of Applied Sciences for Women, University of Delhi India.
Corresponding Author E-mail: manisha.khatri@rajguru.du.ac.in
DOI : http://dx.doi.org/10.13005/bbra/3142

ABSTRACT: Mycobacterium tuberculosis, responsible for causing tuberculosis (TB) in humans, continues to pose a significant worldwide threat, causing extensive fatalities as the most prominent bacterial disease and urgent attention is required to develop novel anti-TB drugs. Throughout the history of medicine, natural remedies have consistently held a vital position, offering valuable references for the development of new drugs. The present study aimed to screen phytoconstituents of Derris indica as inhibitors of protein kinase B, an enzyme critical for cell wall synthesis of Mtb using in silico approach. Molecular docking of phytochemical library of D. indica against PknB was carried out to explore binding interactions, alongwith in silico toxicity prediction of the phytoconstituents. The shortlisted phytoconstituents demonstrated favorable pharmacokinetic characteristics suitable for oral absorption and met the criteria set by Lipinski's rule of five, indicating their potential as drug candidates. Six compounds (Pongaflavanol, Kaempferol, Quercetin, Karanjin, Ovaliflavanone A and Pongaglabrone) demonstrated significant binding interactions with the minimum binding energy ranging from -9.71 kcal/mol to -8.68 kcal/mol as compared with conventional synthetic drugs. These selected phytoconstituents may serve as valuable starting points for the future advancement of effective and safe antimycobacterial drug.
KEYWORDS: ADMET; Antimycobacterial; Drug likeness; Derris indica; Protein Kinase B (PknB); Virtual screening
Download this article as:| Copy the following to cite this article: Kashyap H, Makol V, Khatri M. Screening of Phytochemicals from Derris indica for Antimycobacterial Potential using Molecular Docking Analysis. Biosci Biotech Res Asia 2023;20(3). |
| Copy the following to cite this URL: Kashyap H, Makol V, Khatri M. Screening of Phytochemicals from Derris indica for Antimycobacterial Potential using Molecular Docking Analysis. Biosci Biotech Res Asia 2023;20(3). Available from: https://bit.ly/3Q1ev6W |
Introduction
Tuberculosis (TB) is an infectious disease that can be transmitted from person to person and is a significant contributor to poor health and mortality on a global scale. Before the onset of the COVID-19 pandemic, tuberculosis (TB) occupied the topmost position as the primary cause of mortality attributed to a single infectious agent, surpassing even HIV/AIDS. It is estimated that approximately one-fourth of the world’s population have been exposed to TB, although the majority of individuals will not progress to active TB disease and some will naturally eliminate the infection. Among those who do develop TB each year, around 90% are adults, with a higher prevalence among males compared to females.1
The existing treatment regimen for tuberculosis (TB) is quite extensive, encompassing a prolonged duration and a combination of drugs. However, this approach has drawbacks such as toxic side effects and low patient adherence. Consequently, these challenges have potentially contributed to the emergence of strains which are drug resistant like resistant to multiple drugs and extensively drug-resistant TB (MDR and XDR-TB). These resistant strains have the potential to cause more widespread transmission and larger outbreaks.2 In view of the worldwide TB crisis; there is an urgent need for innovative and efficacious alternatives for treating TB.
A distinguishing characteristic of mycobacteria that differs them from other bacteria is their distinct cell wall structure, which is characterized by species-specific lipids, particularly mycolic acids. As a result, inhibition of enzymes engaged in biosynthesis, transport, and assembly of mycolic acid, have emerged as appealing targets for the design and discovery of novel anti-TB agents.3 PknB (Protein Kinase B) is among the 11 Serine/Threonine protein kinases (STPKs) in bacteria that exhibit a similar structure to those found in eukaryotes. PknB plays a crucial role in various essential bacterial processes, including cell wall synthesis, cell division, and metabolism. Numerous studies have demonstrated that the expression of PknB influences cell morphology and survival with a depletion of PknB resulting in a loss of bacterial viability.4,5 Moreover, it plays a vital role in reactivating cells from a hypoxic state, serving as a critical switch between dormancy and resuscitation.6 Examination of the coding sequence of PknB in 116 clinically isolated drug-resistant strains of M. tuberculosis indicated that the gene is resistant to mutations.7 These factors not only underscore the significance of PknB’s role in Mtb but also emphasize its critical value as a prospective target for drug development.
The abundance of chemically diverse molecules found in natural sources serves as an extensive repository that can offer novel templates for drug design. In recent years, there has been a revived interest in exploring natural sources for the discovery of antimycobacterial/anti-TB agents.8,9,10 Among these natural resources medicinal plants represent a potential option to combat this threat. Derris indica is a plant species found in various regions of Southeast Asia, including Indonesia, Malaysia, and Thailand. For many years, it has been utilized in traditional medicine to address conditions such as bronchitis, whooping cough, rheumatic joints, and to alleviate excessive thirst in diabetes.11 Some researches have shown that crude extracts derived from the stems and roots of D. indica displayed significant antimycobacterial activity against M. tuberculosis H37Ra.12,13 Collins and Frazblau have reported that the crude hexane and dichloromethane extracts from the stems and roots of D. indica exhibited moderate antimycobacterial activity against M. tuberculosis H37Ra with a minimum inhibition concentration (MIC) of 12.5–50 μg/mL.12 The mechanisms by which compounds from D. indica exhibit anti-tuberculosis activity are not yet fully understood. However, some research suggests that these compounds may work by inhibiting the growth and replication of M. tuberculosis within host cells. Additionally, they may also help to stimulate the immune system’s response to the infection, aiding in the clearance of the bacteria from the body.14
This study is an attempt to virtually screen the phytochemicals present in D. indica for their antimycobacterial activity using molecular docking approach. The efficacy of the screened compounds was confirmed through the utilization of in silico docking techniques, by binding to PknB. The compounds were subsequently ranked according to the analysis of their interactions between the protein receptor complex.
Materials and Methods
Database Design
A comprehensive database of phytoconstituents found in D. indica was compiled to facilitate further in silico studies. This database encompassed a wide range of compounds, including organo-sulphur compounds, alkaloids, alcohols, amino acids, flavonoids, fatty acids, fatty acid esters, steroids, carboxylic acids, esters, terpenes, saponins, phenols, and phenolic compounds.
Prediction of drug-likeliness / (ADME analysis)
The estimation of absorption, distribution, metabolism, and excretion (ADME) parameters through in silico methods relies on statistically derived models. In this study, the selected compounds were subjected to evaluation of their ADME parameters using the Swiss online ADME web tool. Additionally, Lipinski’s rule of five for drug-likeness was employed to assess the compounds’ suitability as potential drugs.15,16 The primary purpose of this assay is to offer valuable insights applicable to the process of drug research and development.
Molecular docking
AutoDock 4 and AutoDock tools were used for the in silico docking studies of the selected compounds.17 The crystal structure of Pknb (PDB ID: 2FUM) was retrieved from the protein data bank (www.rcsb.org). Each crystal structure was individually prepared by eliminating existing ligands and water molecules. Missing hydrogens were added to the structure using AutoDock tools. Subsequently, non-polar hydrogens were merged, and polar hydrogens were incorporated into the protein file. The modified structure was saved in a docking-ready PDBQT format. For both standard and test compounds, the 2D chemical structures were obtained from PubChem, and they were further converted into their respective 3D conformations (mol2 format) using the Frog2 server.18 Using AutoDock tools, the protein’s water molecules were eliminated, Kollman charges were introduced, and subsequently, polar hydrogen atoms were added and the ligand molecules were converted and saved into the appropriate format for docking, namely the docking-ready PDBQT format.17 AutoDock 4 was utilized to perform the docking process, where the ligands were docked with PknB, and the binding affinities were subsequently determined.19 The grid center for docking was detected as X = 62.467, Y = 4.815, Z = -32.164 with the dimension of the grid box 30 × 30 × 30. The docking protocol’s accuracy was assessed by docking the reference ligand with PknB and was compared with the docking of the reference activator. The refined protein was used to dock the 3-D optimized ligands, which were then scored using the scoring function. The selection of high-ranking small molecules was based on their binding position and potential interactions with key residues, along with the corresponding binding free energy (ΔG, kcal/mol) values obtained from AutoDock software. Conformations exhibiting the lowest binding affinities (less than -7.0 kcal/mol) were chosen for a closer examination of interacting residues. The docking poses were evaluated using Discovery studio 2021. Furthermore, the top compounds were subjected to evaluation using pharmacokinetics and pharmacodynamics predictors such as SwissADME20 and pkCSM.21
Toxicity Prediction
The potential toxicity of all compounds was predicted through in silico evaluation using the “pkCSM” online computer program.21 In each of these prediction studies, canonical smiles of the compounds were used from the PubChem database. The AMES toxicity, hepatotoxicity, hERG potassium channel inhibition and skin sensitization parameters were predicted to find out the toxicity profile of the identified phytochemical constituents from the D. indica.
Result and Discussion
ADME analysis
Binding of ligands to proteins in an antagonistic manner doesn’t automatically make them desirable as potential drugs. During the drug development process, the assessment of Absorption, Distribution, Metabolism, and Excretion (ADME) is crucial in the early stages of the discovery phase. The influential Lipinski study played a pivotal role in identifying orally active compounds by establishing physicochemical thresholds that are highly indicative of their effectiveness as oral medications, commonly referred to as drug-likeness.22 The Rule-of-Five established a correlation between pharmacokinetic and physicochemical properties. The rule helps to identify molecules that are likely to have good absorption and permeation properties in the human body, which are essential for effective oral drug delivery. As a result, ADME serves as an approach for drug-like testing, enabling informed decisions on whether ligands can be introduced into a biological system.23 These five criterias are generally followed by drug like molecules: molecular weight (Mol. Wt.) up to 500g/mol, Lipophilicity (LogP) to be lower than 5 (milog), the number of donors and acceptors of hydrogen bonds to be lower than 5 and 10 respectively. The idea behind these criteria is that compounds meeting these rules are more likely to have favorable pharmacokinetic properties and are more likely to be orally bioavailable. If a compound fails to meet one or more of these criteria, it may signal the need for additional optimization or consideration of alternative drug delivery routes.
If a drug is intended to be effective when taken orally, it is typically advantageous for the drug to exhibit a lower Topological surface area (TPSA) value. This characteristic improves the drug’s capacity to traverse cell membranes and undergo absorption into the bloodstream. The ADME study results indicated that the selected compounds exhibited favorable pharmacokinetic parameters for oral bioavailability (Table 1) and adhered to Lipinski’s rule of five, indicating drug-likeness. However, compounds that violated two or more criteria of the rule of five were excluded from further study.
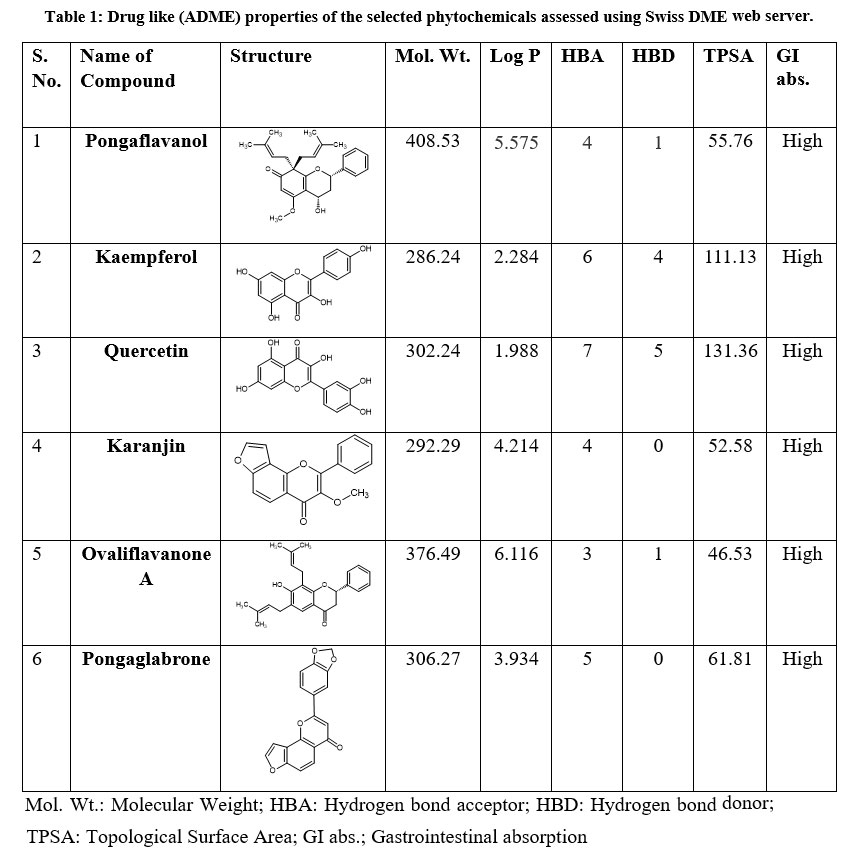 |
Table 1: Drug like (ADME) properties of the selected phytochemicals assessed using Swiss DME web server.
|
Molecular Docking
In silico virtual screening proves to be an effective technique for identifying promising drug-like compounds and to predict the binding affinity of small molecules to a target protein or receptor of interest.24 The current study was conducted to investigate the affinity and binding interactions of the phytochemicals found in D. indica in the active site of protein kinase B (PDB ID : 2FUM) using Autodock in search of anti-mycobacterial compound. To ensure the accuracy of the docking conditions before conducting virtual screening of the Derris constituents, the control inhibitor from its co-crystallized complex was retrieved and re-docked against the respective target using the AutoDock software.
The phytoconstituents were docked in the active site of PknB having ARG35, ASP36, VAL37, ASN67, TYR75, GLU93, VAL95, ASP96, ASP156, and LYS153 amino acid residues. The docked poses of each compound were assessed, and the pose with the lowest binding free energy and the lowest root mean square deviation was selected. Molecular docking scores were determined as the predicted binding free energies in kcal/mol. The lowest binding free energy, representing the best docking score, indicated the highest affinity between the ligand and the protein. Six ligands with the least binding affinity scores are considered for further study. (Table 2) The conventional anti-TB drugs like Isoniazid, Pyrazinamide and Ethambutol were also docked in the active site of PknB to deliberate the anti-TB efficacy of the natural products selected for the study.
Table 2: Docking score (ΔG, kcal/mol) and H-bond interactions of the best docked poses of selected phytochemicals with PknB.
|
S. No. |
Ligand (Compound) |
ΔG (kcal/mol)
|
Number of H-bonds |
Residues involved in H-bonds |
Bond length (Å) |
|
1 |
Pongaflavanol |
-9.71 |
3 |
Asn67, Tyr75 and Asp36 |
2.87, 2.68, 3.09 |
|
2 |
Kaempferol |
-9.07 |
4 |
Asp156, Glu93, Val95, |
2.31, 1.81, 2.25, 1.99 |
|
3 |
Quercetin |
-9.04 |
5 |
Asp156, Glu93, Val95, |
2.30, 1.89, 2.34, 1.98, 2.34 |
|
4 |
Karanjin |
-8.98 |
3 |
Val95, Glu93 |
3.16, 2.87, 2.72 |
|
5 |
Ovaliflavanone A |
-8.84 |
2 |
Glu93, Asn67 |
2.64, 2.73 |
|
6 |
Pongaglabrone |
-8.68 |
2 |
Asp156, Val95 |
2.74, 2.79 |
Among the phytochemicals under study, six compounds (Pongaflavanol, Kaempferol, Quercetin, Karanjin, Ovaliflavanone A and Pongaglabrone) demonstrated significant binding interactions with the minimum binding energy ranging from -9.71 kcal/mol to -8.68 kcal/mol. In comparison the minimum binding energy displayed by the conventional antiTb drugs (Isoniazid -4.84 kcal/mol; Ethambutol -6.89 kcal/mol; Pyrazinamide -4.44 kcal/mol) was found to be very less. The findings suggest that the natural products offer superior potential for treating TB compared to the synthetic drug, which is associated with significant adverse effects. Pongaflavanol showed the strongest binding affinity with the minimum binding energy of -9.71 kcal/mol followed by Kaempferol (-9.07 kcal/mol) among the selected compounds. Figure 1, 2, 3, 4, 5 and 6 illustrates the hydrogen bond interactions between the active phytochemicals and selected amino acid residues for the target protein. Pongaflavanol, a prenylated flavanoid was found to establish three hydrogen bonds with ASN67, TYR75 and ASP36 residues of PknB with bond distances of 2.87, 2.68 and 3.09 respectively (Fig. 1). It was also found to interacting hydrophobically with THR149, SER147, LYS153, GLU93, ARG35 using Vander waals bonding and with TYR94, VAL95 using Pi-Pi stacking. Kaempferol established four hydrogen bonds with the amino acid residues ASP156, GLU93 and VAL95 (Fig. 2) whereas Pongaglabrone made two hydrogen bonds with ASP156 and VAL95 amino acid residue (Fig. 6). Kaempferol extended into the hydrophobic pocket of PknB to establish robust hydrophobic interactions with ALA38, MET155, VAL72, MET92 LEU17, VAL25 whereas Pongaglabrone interacted hydrophobically with LEU17, VAL25, MET92, MET145, ALA38, MET155 using Pi-alkyl binding.
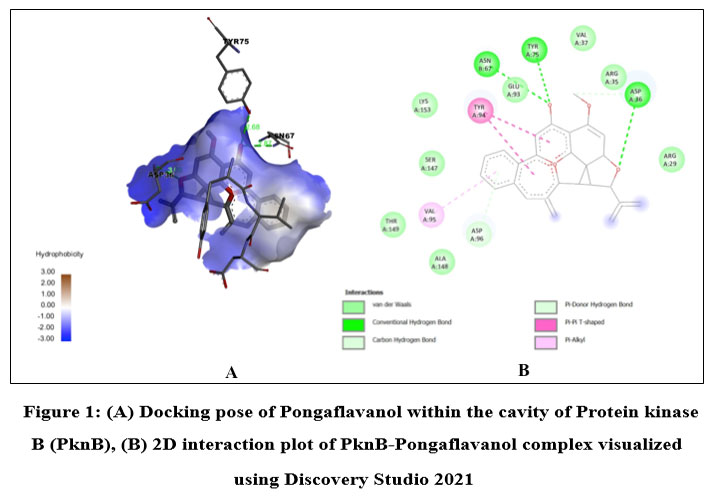 |
Figure 1: (A) Docking pose of Pongaflavanol within the cavity of Protein kinase B (PknB), (B) 2D interaction plot of PknB- Pongaflavanol complex visualized using Discovery Studio 2021
|
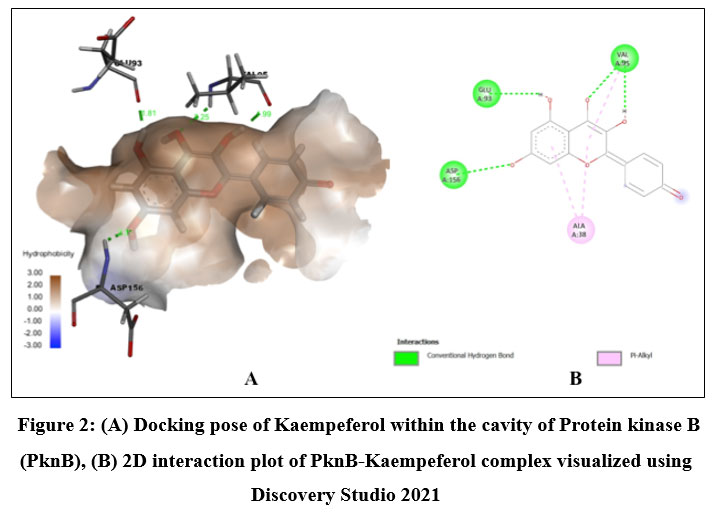 |
Figure 2: (A) Docking pose of Kaempeferol within the cavity of Protein kinase B (PknB), (B) 2D interaction plot of PknB-Kaempeferol complex visualized using Discovery Studio 2021
|
Quercetin, Karanjin and Ovaliflavanone A showed binding affinity with the minimum binding energy -9.04 kcal/mol, -8.98 kcal/mol and -8.84 kcal/mol respectively. Quercetin, a flavanoid commonly found in fruits, vegetables and grains interacted with ASP156, GLU93 and VAL95 using five hydrogen bonds. Hydrophobically Quercetin was found to be interacting with TYR94, MET145, LEU17, MET92, MET155, VAL72 and ALA38 (Fig. 3). Karanjin, a bioactive furanoflavonoid and a potent biomolecule25 established three hydrogen bonds with VAL95 and GLU93 (Fig. 4) whereas Ovaliflavanone A interacted using two hydrogen bonds with GLU93 and ASN67 amino acid residue (Fig. 5). These molecular docking studies therefore helped in establishing that these phytochemicals from Derris indica have the potential to serve as antimycobacterial compounds.
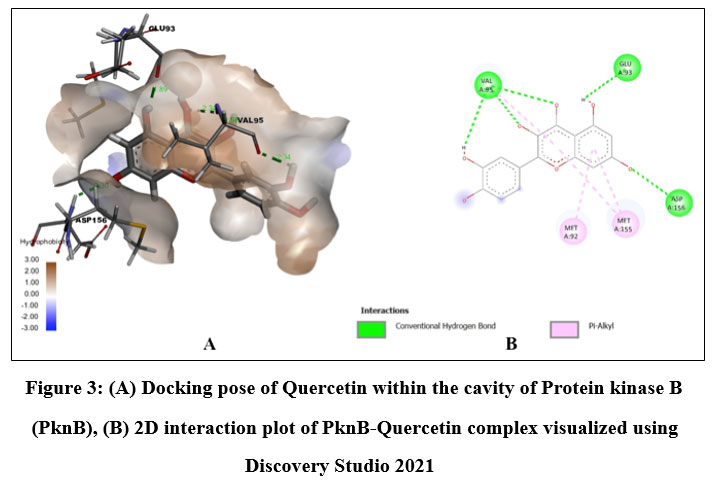 |
Figure 3: (A) Docking pose of Quercetin within the cavity of Protein kinase B (PknB), (B) 2D interaction plot of PknB-Quercetin complex visualized using Discovery Studio 2021
|
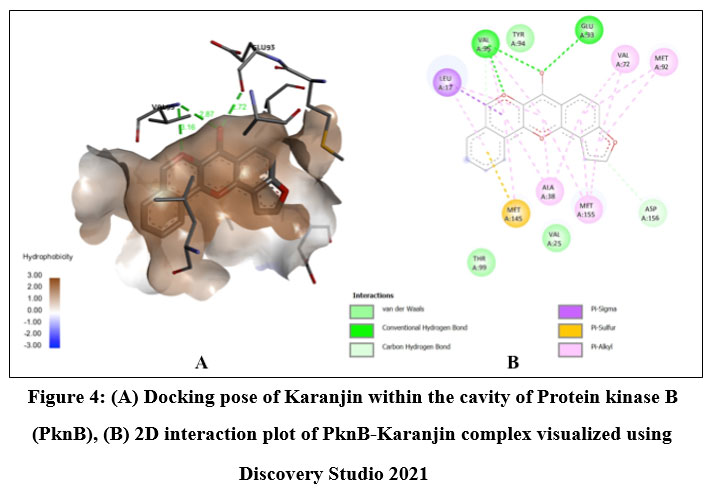 |
Figure 4: (A) Docking pose of Karanjin within the cavity of Protein kinase B (PknB), (B) 2D interaction plot of PknB- Karanjin complex visualized using Discovery Studio 2021.
|
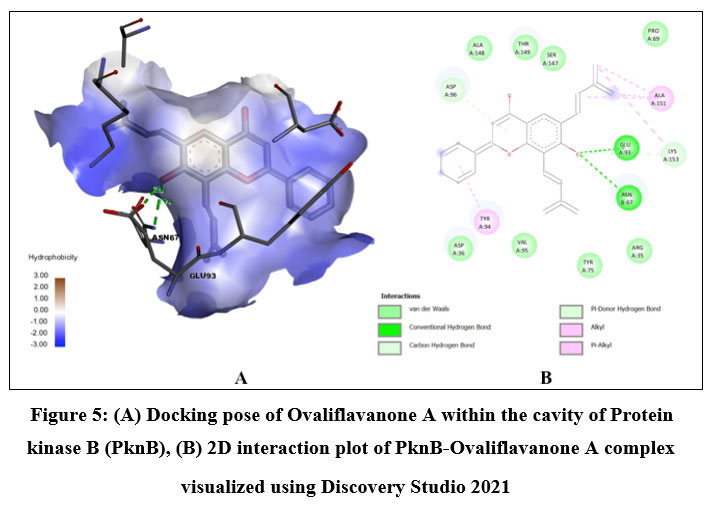 |
Figure 5: (A) Docking pose of Ovaliflavanone A within the cavity of Protein kinase B (PknB), (B) 2D interaction plot of PknB-Ovaliflavanone A complex visualized using Discovery Studio 2021
|
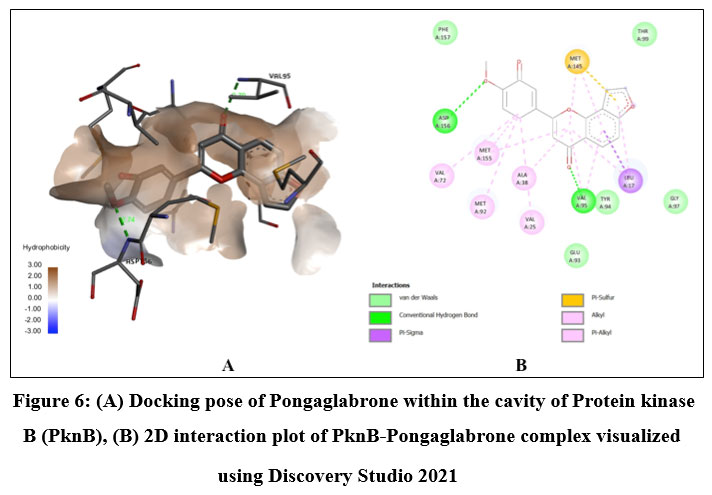 |
Figure 6: (A) Docking pose of Pongaglabrone within the cavity of Protein kinase B (PknB), (B) 2D interaction plot of PknB-Pongaglabrone complex visualized using Discovery Studio 2021
|
In silico molecular docking approaches have significantly advanced drug discovery and development, but they also come with certain limitations. In silico methods often simplify the intricate biological processes and interactions that occur in living organisms, leading to potential inaccuracies in predictions. Integrating these methods with experimental validation and a thorough understanding of their scope and potential pitfalls is crucial for leveraging their benefits effectively.
Prediction of toxicity
The potential toxicity of the selected compounds, including mutagenicity, cardiotoxicity, carcinogenicity, skin irritation, immunotoxicity, and reproductive toxicity, was assessed using the pkCSM online tool (Table 3). pkCSM is an accessible web server that utilizes graph-based signatures to provide a comprehensive platform for efficiently evaluating the pharmacokinetic and toxicity properties of small molecules.21 Predicting toxicity using the AMES test plays a crucial role in identifying mutagenic compounds early in drug discovery. Although most of the compounds were predicted negative but Karanjin and Pongaglabrone were found positive for AMES toxicity/ Mutagenicity. Blockers of the Ether-À-Go-Go-Related Gene (HERG potassium K+) channel have the potential to induce toxicity, and the forecasted IC50 value frequently offers a reasonably accurate prediction regarding the cardiac toxicity of drugs during the initial phases of development. Cardiac toxicity (hERG II inhibition) was not observed in any of the selected compounds. Predicting hepatotoxicity using online tools helps identify potential liver toxicity issues in the early stages, enabling researchers to make informed decisions about compound progression. Ensuring a drug’s safety profile extends to its effect on the skin is essential for patient well-being. Hepatotoxicity was predicted for Pongaflavanol whereas skin toxicity was not observed in any of the compounds. Human responses to drugs can vary due to genetic, environmental, and physiological factors. Online tools might not effectively address individual variability, leading to inaccurate predictions for certain populations. In this context, the preliminary assessment conducted in silico can complement future studies regarding the safety of these compounds, however they should be used in conjunction with experimental data.
Table 3: Toxicity of the selected phytoconstituents assessed using the SwissADME web server.
|
S.No |
Name of Compound |
AMES toxicity |
Maximum tolerated dose (mg/kg/day) |
hERG I inhibition |
hERG II inhibition |
Oral rat acute toxicity LD50 (mol/kg) |
Oral rat chronic toxicity |
Hepato-toxicity |
Skin toxicity |
|
1 |
Pongaflavanol |
No |
0.075 |
No |
No |
2.586 |
1.941 |
Yes |
No |
|
2 |
Kaempferol |
No |
0.935 |
No |
No |
2.329 |
2.616 |
No |
No |
|
3 |
Quercetin |
No |
0.779 |
No |
No |
2.513 |
2.636 |
No |
No |
|
4 |
Karanjin |
Yes |
0.021 |
No |
No |
2.399 |
1.641 |
No |
No |
|
5 |
Ovaliflavanone A |
No |
0.447 |
No |
No |
2.126 |
1.945 |
No |
No |
|
6 |
Pongaglabrone |
Yes |
0.352 |
No |
No |
2.328 |
1.474 |
No |
No |
AMES toxicity: Carcinogenic (or Mutagenic) potential; hERG I & hERG II inhibition: cardio-toxicity; Oral rat chronic toxicity: log mg/kg_bw/day; Skin toxicity: Sensitization of Skin.
Conclusion
In this current study, Phytochemicals from the plant Derris indica were explored for their antimycobacterial potential using molecular docking against protein kinase B (PknB) enzyme, one of the therapeutic targets of Tuberculosis. All the compounds were investigated for their drug likeness, ADMET prediction and then docked with the enzyme. Further investigation led to six compounds (Pongaflavanol, Kaempferol, Quercetin, Karanjin, Ovaliflavanone A and Pongaglabrone) showing highest binding affinity with the minimum binding energy ranging from -9.71 kcal/mol to -8.68 kcal/mol in comparison with conventional antiTB drugs. No cardiotoxicity and skin toxicity was predicted in any of the compounds. Therefore, these natural compounds have the potential to become valuable leads in the development of antimycobacterial drugs that are both clinically effective and safe. However, it is necessary to conduct further studies and make structural modifications to these phytoconstituents in order to create antimycobacterial drugs that are both safe and efficient.
Acknowledgments
The authors are thankful to Shaheed Rajguru College of Applied Sciences, University of Delhi, for providing the research facility to conduct the study.
Conflict of Interest
There is no conflict of interest.
Funding Sources
There are no funding sources.
References
- World Health Organization. 2022. Global tuberculosis report.
- Koul A., Arnoult E., Lounis N., Guillemont J., and Andries K. The challenge of new drug discovery for tuberculosis. Nature. 2011;469:483–490.
CrossRef - Nguyen-Hung L., Marie L. P., Alexandre S., Nicolas T., Virginie ., Odile B. Z., Mamadou D., and Hedia M., The protein kinase PknB negatively regulates biosynthesis and trafficking of mycolic acids in mycobacteria, Journal of Lipid Research. 2020;61:1180-1191.
CrossRef - Kang, C. M., Abbott D., Park S.T., Dascher C.C., Cantley L., and Husson R.N. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: Substrate identification and regulation of cell shape. Genes Dev. 2005;19:1692–1704.
CrossRef - Manuse S., Fleurie A., Zucchini L., Lesterlin C., and Grangeasse C. Role of eukaryotic-like serine/threonine kinases in bacterial cell division and morphogenesis. FEMS Microbiol. Rev. 2015;40: 41–56.
CrossRef - Emane A.K.A., Guo X., Takiff H.E., and Liu S. Drug resistance, fitness and compensatory mutations in Mycobacterium tuberculosis. 2021;129:102091.
CrossRef - Gupta A., Pal S. K., Pandey D., Fakir N. A., and Rathod, S. PknB remains an essential and a conserved target for drug development in susceptible and MDR strains of tuberculosis. Ann. Clin. Microbiol. Antimicrob.2017;16: 56.
CrossRef - Guzman J.D., Gupta A., Bucar F., Gibbons S., and Bhakta S. Antimycobacterials from natural sources: ancient times, antibiotic era and novel scaffolds. Front Biosci. 2012;17(5):1861–81.
CrossRef - Dashti Y., Grkovic T., and Quinn R.J. Predicting natural product value, an exploration of anti-TB drug space. Nat Prod Rep. 2014;31(8):990–8.
CrossRef - Santhosh R.S., and Suriyanarayanan B. Plants: a source for new antimycobacterial drugs. Planta Med. 2014;80(1):9–21.
CrossRef - Yadav P.P., Ahmad G., and Maurya R. Furanoflavonoids from Pongamia pinnata fruits. Phytochemistry 2004;65:439–443.
CrossRef - Collins L., and Franzblau S.G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Anti- microb. Agents Chemother. 1997;41:1004–1009.
CrossRef - Koysomboon S., Altena I.V., Kato S., and Chantrapromma K. Antimycobacterial flavonoids from Derris indica, Phytochemistry 2006;67:1034–1040
CrossRef - Muqarrabun L.M.R. A., Ahmat N., Ruzaina S.A.S., Ismail N.H., and Sahidin I., Medicinal uses, phytochemistry and pharmacology of Pongamia pinnata (L.) Pierre: A review, Journal of Ethnopharmacology. 2013;150:395–420.
CrossRef - Gleeson M.P., Hersey A., and Hannongbua S. In-silico ADME models: a general assessment of their utility in drug discovery applications. Top. Med. Chem. 2011;11(4):358-381.
CrossRef - Lipinski CA., Lead and drug-like compounds the rule-of- five revolution. In Drug Discovery Today: Technologies. 2004.
CrossRef - Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., and Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Comput. Chem. 2009;30(16):2785-2791.
CrossRef - Miteva M.A., Guyon F., and Tufféry P. Frog2: Efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res. 2010;38:622-W627.
CrossRef - Trott O., and Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Comput. Chem. 2010;31(2):455-461.
CrossRef - Daina A., Michielin O., and Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Rep. 2017;7:42717.
CrossRef - Pires D.E.V., Blundell T.L., and Ascher D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using
Graph-Based Signatures. Med. Chem. 2015;58:4066–4072.
CrossRef - Lipinski C.A., Lombardo F., Dominy B.W., and Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Drug Deliv. Rev. 2001;46(1-3):3-26.
CrossRef - Gleeson M.P., Hersey A., and Hannongbua S. In-silico ADME models: a general assessment of their utility in drug discovery applications. Top. Med. Chem. 2011;11(4):358-381.
CrossRef - Meng X.Y., Zhang H.X., Mezei M., and Cui M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Comput. Aided Drug Des.2011;7:146–157.
CrossRef - Noor A.A.M., Othman S.N.N., Lum P.T., Mani S., Shaikh M.F., and Sekar M. Molecules of interest–Karanjin–A review. J.2020;12.
CrossRef - Saeidnia S., Manayi A., and Abdollahi M. The Pros and Cons of the In siliso Pharmaco-toxicology in Drug Discovery and Development. Int J Pharm. 2013;9:176-181.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.



