

How to Cite | Publication History | PlumX Article Matrix
Cardiotoxic Drugs: An Insight into its Pathologic Mechanisms
Anisha Sara Anil , Sonale S and N Venkateswaramurthy *
, Sonale S and N Venkateswaramurthy *
Department of Pharmacy Practice, J. K. K. Nattraja College of Pharmacy, Kumarapalayam, Tamil Nadu, India.
Corresponding Author E-mail: nvmurthi@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3201

ABSTRACT: Cardiovascular diseases are among the major causes of mortality and morbidity worldwide Cardiotoxicity due to drugs is a common and significant adverse effect on cardiovascular health, acting through multifactorial pathological mechanisms. Drug-induced cardiotoxicity limits the use and further development of certain drugs. Keeping this in mind, this review discusses the crucial drug-receptor interactions involved in cardiotoxicity induced by some drugs such as cocaine, trastuzumab, isoproterenol, antidiabetic drugs like pioglitazone, theophylline, ergotamine, methysergide, anthracyclines, fluoropyrimidines, cisplatin, NSAIDs, and antiviral agents. The key receptors involved in the pathological mechanism behind the cardiotoxicity induced by these drugs are discussed, aiming to provide in-depth knowledge for future drug discovery and prevention of drug-induced cardiotoxicity.
KEYWORDS: Antivirals; Antineoplastics; Cardiotoxicity; Cardiotoxic drugs; Cocaine; Isopreterenol; NSAIDs; Theophylline
Download this article as:| Copy the following to cite this article: Anil A. S, Sonale S, Venkateswarsamurthy N. Cardiotoxic Drugs: An Insight into its Pathologic Mechanisms. Biotech Res Asia 2024;21(1). |
| Copy the following to cite this URL: Anil A. S, Sonale S, Venkateswarsamurthy N. Cardiotoxic Drugs: An Insight into its Pathologic Mechanisms. Biotech Res Asia 2024;21(1). Available from: https://bit.ly/3uwoSYn |
Introduction
Cardiovascular diseases are considered the leading cause of morbidity and mortality in nearly all developed countries worldwide. Therefore, the negative impact of drugs or toxins on the cardiovascular system must not be understated.1-4 Cardiotoxicity is commonly used to describe toxicity that negatively impacts the heart and may lead to conditions such as arrhythmia, myocardial infarction, and myocardial hypertrophy.5 Generally, cardiotoxicity results from the concurrent disruption of the principal functions and viability of the myocardium.6 Drug-induced cardiotoxicity is a clinically significant issue, as it can lead to both cardiac dysfunction and myocardial injury. Over the last few decades, cardiovascular side effects have forced the withdrawal of more than 10% of clinical medications from the market, hindering future drug development and negatively impacting patient health advancement.7
Between 1994 and 2006, 45% of all withdrawn medications were due to cardiotoxicity, primarily caused by adverse effects associated with cardiac ischemia and arrhythmogenicity.8 Drug-induced cardiotoxicity restricts or prohibits further use of the specified drug. One of the main reasons for removing drugs from clinical trials and the market is concern over cardiac safety.6 Consequently, drug-induced cardiotoxicity significantly restricts drug research and the clinical management of already approved medications.9 The likelihood of cardiotoxic symptoms is influenced by various factors, including medication types and actions, pharmacokinetics, underlying heart diseases, genetic factors, and other conditions that may affect a drug’s response when administered at high doses.10
Numerous drug classifications, such as anti-cancer, anti-diabetics, anti-viral, etc., can lead to undesirable cardiovascular consequences. This review summarizes the drug-receptor mechanism by which drug-induced cardiotoxicity may develop. It covers the cardiotoxic mechanisms of some clinically used drugs, such as cocaine, trastuzumab, isoproterenol, antidiabetic drugs like pioglitazone, theophylline, ergotamine, methysergide, anthracyclines, fluoropyrimidines, cisplatin, NSAIDs, and antiviral agents. The review aims to enhance the current understanding of the pathology behind the cardiovascular toxicities of these drugs.
Pathophysiological Mechanism of Cardiotoxic Drugs on Cardiovascular Health
Cocaine
Cocaine is the most frequently exploited recreational drug due to the stimulant and euphoric effects it exerts on the brain and central nervous system11 Derived from the leaves of the South American Andean shrub Erythroxylon coca, cocaine is a tropane alkaloid substance. First utilized as an anaesthetic agent for local surgeries in the 1880s, it became a popular recreational drug by the 1970s. Cocaine is one of the most commonly used recreational drugs globally, with an estimated 18 million users worldwide.12In the United States, cocaine use is a significant cause of morbidity and mortality, with consequences ranging from long-term cognitive decline to early death.13-16
Cocaine usage is associated with numerous acute and chronic cardiovascular consequences, including aortic dissection, ischemia, hemorrhagic stroke, chest pain, hypertension, acute myocardial infarction (MI), arrhythmias, sudden death, and cardiomyopathy leading to chronic heart failure17,18 (Figure1). The primary cause of cocaine’s cardiovascular (CVS) damage is its significant sympathomimetic action. Increased alpha-1 adrenergic stimulation promotes arterial vasoconstriction, leading to a rise in blood pressure and a reduction in microvascular blood flow, while increased beta-adrenergic stimulation enhances cardiac contractility and heart rate.19 Additionally, cocaine enhances the release of the potent vasoconstrictor endothelin-1, alongside the α-adrenergic receptors of the smooth muscle cells in the coronary arteries, and inhibits the synthesis of the vasodilator nitric oxide.17 Nitric oxide and endothelin-1 are natural rivals for maintaining vascular function. An imbalance created by stimulated endothelin-1 and inhibited nitric oxide can lead to vasoconstriction, increased blood pressure, and potential vascular remodeling and dysfunction.20
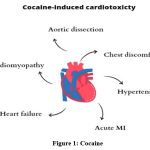 |
Figure 1: Cocaine |
Trastuzumab
Trastuzumab is a humanized monoclonal antibody (mAb) that targets the Human Epidermal Growth Factor Receptor-2 (HER-2), also known as the Erythroblastic Leukemia Viral Oncogene Homolog 2 (ErbB2).22 Approved for use in the US in 1998, Trastuzumab is indicated for the treatment of metastatic breast cancer in female patients whose tumors overexpress the HER-2 protein.5 While this medication is expected to be less hazardous to other cells in the body due to its targeted approach at tumor cells, cardiotoxicity with this drug was quickly noted in clinical settings.22
Trastuzumab has been linked to cardiotoxicity, including heart failure and cardiac insufficiency. Ongoing research seeks to elucidate the pathophysiology of cardiotoxicity associated with Trastuzumab. Recent case reports indicate the presence of HER-2 in human cardiac tissue,23 which plays a crucial role in embryonic cardiogenesis and cardiac hypertrophy. 22 It is possible that Trastuzumab directly damages the heart, potentially through the cardiac HER-2 receptor. Under physiological conditions, Neuregulin-1 interacts with Epidermal Growth Factor Receptor-2 (ErbB2) to promote the formation of an ErbB4/ErbB2 heterodimer that prevents cell death in cardiomyocytes via AKT-dependent signaling. 24
Trastuzumab acts as an inhibitor of ErbB2, preventing ErbB4/ErbB2 heterodimerization and consequently inducing apoptosis.25 The interaction between Trastuzumab and ErbB2 activates downstream signal transduction pathways, leading to increased Bax and Bcl-xS levels, decreased levels of Bcl-xL, and activation of the caspase cascade. Moreover, the reduction in mitochondrial membrane potential (MMP) caused by the suppression of Bcl-xL and the ensuing mitochondrial energy crisis contribute to Trastuzumab-induced apoptosis.26 It is hypothesized that interaction with cardiac cells pathologically overexpressing HER-2 is responsible for Trastuzumab-associated cardiotoxicity27 (Figure 2).
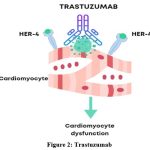 |
Figure 2: Trastuzumab |
Isoproterenol (Isoprenaline)
Isoproterenol, also known as isoprenaline, is a medication used to treat bradycardia (slow heart rate). Structurally similar to epinephrine, it was first approved for use in the US in 1947.28 Isoproterenol has been utilized as a model compound to induce infarct-like lesions in rats and several other animals.29 The changes in the myocardium induced by isoproterenol resemble, in some aspects, those occurring in human myocardial infarction.30
It is believed that isoproterenol’s adrenergic cardiostimulatory activity causes cardiac oxidative metabolism to increase to a level that surpasses the oxygen supply available to the myocyte through the coronary circulation. This energy imbalance, coupled with intricate biochemical (such as altered calcium influx, activation of the adenyl cyclase system, platelet aggregation, and production of Reactive Oxygen Species)31 and structural changes (including alterations in membrane permeability),32 appears to play a role in the pathogenesis of myocyte damage.29 The complex mechanisms of cardiotoxicity involve overstimulation of beta-adrenoceptors and the generation of Reactive Oxygen Species. When β-adrenoceptors are overstimulated, myocardial energy demands increase. Additionally, due to its potent β2-adrenoceptor agonist effects, isoprenaline causes significant peripheral vasodilation, which substantially lowers diastolic blood pressure and consequently decreases myocardial perfusion.33-35 (Figure 3)
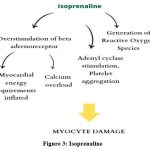 |
Figure 3: Isoprenaline |
Another common outcome of β1-adrenoceptor overstimulation is calcium overload.36-38 Toxicity is also likely influenced by increased platelet aggregation.39-41 High quantities of isoprenaline can lead to the direct formation of Reactive Oxygen Species42-46 through metal-catalyzed oxidation or spontaneously due to ischemia. Given the complex pathophysiology, no single medication can completely prevent or reverse the damage caused by isoprenaline, and some only act at lower doses of the drug.41,47-49
Pioglitazone
Pioglitazone is an antihyperglycemic medication that enhances hepatic and peripheral insulin sensitivity in cases of insulin resistance. It inhibits hepatic gluconeogenesis and increases peripheral and splanchnic glucose uptake.50Approved in 1999 for improving glycemic control in people with type 2 diabetes mellitus, Pioglitazone followed the introduction of the first glitazone class drug, troglitazone, in the USA in March 1997.51 As first-line treatments, rosiglitazone and pioglitazone were introduced to the American market in 1999, to be taken either alone or in conjunction with other medications.52 Currently, only Pioglitazone is available for use, either alone or combined with metformin or sulphonylureas. Initially, thiazolidinedione anti-diabetic medications, like rosiglitazone and pioglitazone, were considered protective against diabetic cardiomyopathy and ischemia-reperfusion injury.53 However, with their clinical applications being expanded, they have been found to have detrimental side effects, such as cardiac failure and myocardial hypertrophy.54
The cardiovascular effects of Pioglitazone may be linked to the regulation of angiogenesis, neointima formation, and atherosclerosis associated with VEGFR-2-participating pathways. Research indicates that Pioglitazone significantly binds to VEGFR-2.55-57 The tyrosine kinase receptor VEGFR-2, activated by trans-phosphorylation, dimerizes in response to ligand interaction.57 The activation of VEGFR-2 triggers the c-Raf/MEK/ERK and PI3K/Akt pathways, which increase cell proliferation, migration, and survival.57,58 The hypertrophic growth of cardiomyocytes is dependent on VEGFR-2.59,60
Pioglitazone specifically targets VEGFR-2 and reduces phospho-VEGFR-2 expression, suggesting that by blocking VEGFR-2 signaling, Pioglitazone promotes cardiomyocyte apoptosis and prevents cardiomyocyte hypertrophy in neonatal rats. The downstream PI3K/Akt signaling pathway also supports these cells’ survival and hypertrophy, functioning by suppressing P53-dependent pathways and stimulating mTOR-dependent pathways.61
Theophylline
Theophylline (1,3-dimethylxanthine) is predominantly used as a bronchodilator for patients with asthma and chronic obstructive pulmonary disease (COPD) worldwide. In the United States, while asthma and COPD are primarily treated with other medications, Theophylline is mostly used to treat bradycardia and apnea in premature neonates. It triggers the endogenous release of catecholamines through indirect activation of beta-1 and beta-2 receptors, resulting in desirable bronchodilation at therapeutic doses. Unfortunately, Theophylline has a narrow therapeutic window, and concentrations slightly above this range can lead to a variety of adverse effects in the context of acute and chronic toxicity.62
Theophylline toxicity is linked to significant clinical symptoms caused by high levels of circulating catecholamines. Toxicity occurs when serum Theophylline levels exceed the therapeutic range, which can happen either intentionally through overdose or unintentionally when Theophylline metabolism and/or clearance are altered due to certain physiological stresses.62 Theophylline blocks adenosine receptors, leading to both beneficial effects, such as bronchodilation, and harmful effects, including tachycardia, cardiac arrhythmias, seizures, and cerebral vasoconstriction.1 Although the exact mechanism is complex, it clearly involves antagonistic activity at adenosine receptors. Theophylline, along with a small amount of caffeine, causes the release of catecholamines through an unidentified mechanism.1,63
At higher doses, Theophylline inhibits phosphodiesterase, thereby increasing levels of cyclic adenosine monophosphate, which further increases adrenergic activation and catecholamine release. In cases of Theophylline toxicity, epinephrine levels can be 4- to 8-times higher than normal, while norepinephrine concentrations can be 4- to 10-times greater. Elevated catecholamine levels can lead to various adverse effects, including cardiovascular arrhythmias, metabolic acidosis, hyperglycemia, and hypokalemia.62
Ergotamine and Methysergide
Ergot compounds, such as Methysergide and ergotamine, are commonly used to treat migraines. Ergotamine acts as an alpha-adrenergic blocker with serotonin antagonistic properties and directly stimulates the smooth muscle of peripheral and cerebral blood vessels. Methysergide, on the other hand, is a central 5-hydroxytryptamine (5-HT) agonist, particularly at therapeutic nuclei, but also acts as a powerful peripheral 5-HT inhibitor, exhibiting competitive blockade of vascular 5-HT receptors. Several case studies have linked both medications to valvular defects involving the mitral, aortic, and tricuspid valves, occasionally resulting in right-sided heart failure.64-66
Anthracyclines
Since the 1960s, anthracyclines (ANTs), such as doxorubicin, epirubicin, and daunorubicin, have been recognized as the prototype of type 1 cardiotoxicity (CTX).67 ANTs can cause irreversible type 1 CTX by producing reactive oxygen and nitrogen species (ROS and RNS).68-70 A key mechanism by which ANTs induce CTX is their interaction with topoisomerase 2 (TOP2) A and -B, which is abundantly expressed in cardiomyocytes. Additionally, drugs like doxorubicin not only directly harm cardiomyocytes but also promote apoptosis in immune (e.g., macrophages) and cancer cells, leading to the release of high mobility group box 1 (HMGB1), which activates toll-like receptors (TLR)-2 and -4.71,72 Cardiac dysfunction following ANT therapy is associated with higher mitochondrial iron levels compared to normal hearts. This suggests that iron accumulation in the mitochondria and oxidative stress are significant contributors to ANT-induced CTX.73 Furthermore, anthracyclines disrupt mitochondrial structural integrity and impair cardiac mitochondrial function. Doxorubicin, for example, has been shown to induce receptor-interacting protein 3 (RIPK3)-induced activation of Ca2+-calmodulin-dependent protein kinase (CaMKII), which leads to the opening of the mitochondrial permeability transition pore (MTPT), culminating in necroptotic cardiomyocyte death.74 (Figure 4)
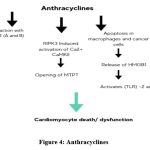 |
Figure 4: Anthracyclines |
Antimetabolites
Fluoropyrimidines, such as gemcitabine, capecitabine, and 5-fluorouracil (5-FU), are used to treat various tumors. While the cardiovascular side effects of fluoropyrimidines are generally reversible, they can cause cardiomyocyte death and loss. This occurs through coronary artery thrombosis and myocardial infarction, as well as directly through cardiomyocyte-intrinsic mechanisms. These medications also induce coronary artery spasm and myocardial ischemia.5,75-77 The cardiotoxicity (CTX) of 5-FU and its metabolites is attributed to several factors, including the suppression of nitric oxide (NO),78,79 increased production of reactive oxygen and nitrogen species (ROS/RNS),80 enhanced endothelium thrombogenicity,81 senescence,82 and DNA and RNA damage. Cardiomyocytes and endothelial cells are particularly vulnerable to oxidative stress triggered by 5-FU. The drug upregulates endothelin 1, dysregulates endothelial nitric oxide synthase (eNOS), and activates protein kinase C. These effects result in coronary spasm and both endothelium-dependent and -independent vasoconstriction.83,84
Alkylating Drugs – Cisplatin
Cisplatin, a broad-spectrum chemotherapeutic medication belonging to the alkylating group, is used to treat various tumor types, including sarcomas, carcinomas (such as small cell lung cancer and ovarian cancer), lymphomas, and germ cell tumors. Cardiovascular disorders, notably myocardial infarction and angina, have been linked to cisplatin-based chemotherapy in 7–32% of patients.85 Cisplatin-induced heart failure is associated with depolarization of the mitochondrial membrane and alterations in mitochondrial ultrastructure. Additionally, cardiomyocytes show signs of endoplasmic reticulum stress response activation, increased caspase 3 activity, and an accelerated rate of apoptosis following cisplatin administration.86
NSAIDs
Despite being among the most frequently prescribed and used medications worldwide, nonsteroidal anti-inflammatory drugs (NSAIDs) are associated with several significant, and sometimes fatal, adverse drug reactions (ADRs).87 The inhibition of COX-2 by traditional NSAIDs (tNSAIDs) and coxibs can reduce the production of prostacyclin (PGI2) by endothelial cells, a crucial vasodilator and platelet inhibitor. However, this inhibition does not affect the production of COX-1-derived thromboxane A2 (TXA2), a vasoconstrictive eicosanoid that promotes platelet aggregation and thrombus formation. This imbalance between PGI2 and TXA2 increases the risk of atherosclerosis and cardiovascular thrombotic events.88,89
Selective COX-2 inhibition may also directly affect the production of PGI2 and prostaglandin E2 (PGE2) in the heart. Beyond their role in inhibiting the cyclooxygenase pathway, NSAIDs have been shown to induce cell death by inhibiting the Akt signaling pathway,90 downregulating the NF-κB pathway,91 upregulating the nonsteroidal activated gene-1,92 and altering the p53 pathway,93 all of which are implicated in apoptosis.94 Additionally, the suppression of COX by NSAIDs leads to the accumulation of arachidonic acid, which inhibits the phosphorylation of cardiac mitochondria. This results in increased production of reactive oxygen species (ROS) and decreased activity of antioxidant enzymes, leading to oxidative stress that contributes to heart failure.95,96
Remdesivir
Remdesivir is a prodrug of the nucleoside triphosphate metabolite, which originates from the nucleotide analogue GS443902, an adenosine nucleotide. Adenosine is known to exhibit antiarrhythmic effects in atrioventricular (AV) re-entrant tachycardias by causing temporary AV nodal block. However, in individuals with structural heart disease, it may induce proarrhythmia.97
Adenosine is also a potent vasodilator, which can lead to severe hypotension and the subsequent release of catecholamines. These events have the potential to evolve into ventricular fibrillation (VF) or ventricular tachycardia (VT).Additionally, adenosine can shorten the atrial action potential, increase atrial refractoriness, and potentially cause atrial fibrillation (AF). These effects may also extend to ventricular cells, putting individuals at risk for VF. Importantly, adenosine can cause spatial and temporal heterogeneity in ventricular refractoriness, which may result in VF. This highlights the complexities associated with the cardiovascular effects of Remdesivir and its metabolites.98
Table 1
| S.No | Drugs | Mechanism of action | Cardiotoxic Effects |
| 1. | Cocaine | Increase alpha 1 and beta adrenergic Stimulation. | Vasoconstriction, Increased blood pressure, Aortic dissection, Cardiomyopathy. |
| 2. | Trastuzumab | Prevents ErbB2/ErbB4 heterodimerization. | Induced cardiac cells apoptosis. |
| 3. | Isoproterenol | Overstimulation of beta adrenoreceptors and the generation of reactive oxygen species. | Myocyte damage |
| 4. | Pioglitazone | Targets VEGFR-2 and decreases phosphor-VEGFR-2 expression. | Cardiomyocyte apoptosis |
| 5. | Theophylline | Blocks adenosine receptors | Tachycardia, cardiac arrhythmias. |
| 6. | Ergotamine and Methysergide | Ergotamine is alpha- adrenergic blocker with serotonin antagonistic characteristics.
Methysergide does competitive blockage of vascular 5- hydroxy tryptamine receptors. |
Mitral, aortic, and tricuspid valve defects. |
| 7. | Anthracyclines | Cause irreversible type 1 CTX by producing reactive oxygen and nitrogen species.
interaction with topoisomerase 2 (TOP2) A and -B |
Cardiomyocyte death |
| 8. | Antimetabolites | The suppression of NO, increased ROS/RNS production, greater endothelium thrombogenicity and DNA and RNA damage. | Coronary artery spasm and myocardial ischemia |
| 9. | Alkylating Agents | CISPLATIN (Depolarization of the mitochondrial membrane and changes in the ultrastructure of the mitochondria. | Heart failure. |
| 10. | NSAIDS | Inhibits the Akt signaling pathway, downregulates the NF- b pathway, upregulates the nonsteroidal activated gene-1, and alter the p53 pathway. | Cardiac cell apoptosis |
| 11. | Antiviral Agents | REMDESIVIR (Cause temporary AV nodal block). | Antiarrhythmic effects. |
Management of Drug-Induced Cardiotoxicity
Managing drug-induced cardiotoxicity requires a multifaceted approach tailored to the condition’s severity. Prompt discontinuation of the offending drug is crucial, followed by the administration of antidotes and supportive therapies. Experts recommend initiating treatment with primary heart failure medications and seeking early cardiology consultation for symptoms or signs of trastuzumab-induced cardiotoxicity.99 According to ACC/AHA guidelines, first-line agents include ACE inhibitors (ACEIs), angiotensin receptor blockers (ARBs), and beta-blockers (BBs), followed by loop diuretics, hydralazine-nitrates, and aldosterone antagonists.100 Additional newer agents, such as sacubitril-valsartan, a neprilysin-angiotensin receptor antagonist, and ivabradine, an If channel inhibitor, may also be beneficial.101
In cases of advanced heart failure, potential therapeutic options include cardiac resynchronization therapy, mechanical circulatory support, and orthotopic heart transplant.102 Carvedilol, a dual beta and alpha-1 blocker with robust antioxidant capabilities, has shown effectiveness in preventing left ventricular dysfunction induced by anthracyclines and/or trastuzumab. This benefit is achieved by mitigating reactive oxygen species formation, correcting mitochondrial abnormalities, and reducing cardiomyocyte apoptosis. The concurrent use of enalapril and carvedilol appears advantageous in managing cardiotoxicity induced by anthracyclines without altering the drugs’ intended therapeutic effects.103
Remote ischemic conditioning, a noninvasive and nonpharmacological intervention using a blood pressure cuff to create brief episodes of ischemia and reperfusion in a peripheral limb, is being explored as a cardioprotective treatment for cancer patients. Its exact mechanism involves humoral and neural pathways and is believed to activate pro-survival pathways that regulate mechanisms implicated in ischemia-reperfusion injury and anthracycline cardiotoxicity, such as calcium overload, lipid peroxidation, ROS generation, and mitochondrial modulation.104
Fenofibrate and PEG-SOD prevent cardiac dysfunction induced by doxorubicin by normalizing oxidative stress and modulating NF-kappaB signaling.105 Limiting the cumulative dose of cytotoxic drugs and adding dexrazoxane can help protect against cardiotoxicity.106 Early detection and management of anthracycline-induced cardiotoxicity through biomarkers and angiotensin-converting enzyme inhibitors can avert heart failure development, enhancing patient quality of life.107 Dexrazoxane prevents doxorubicin cardiotoxicity by reducing Top2beta-mediated DNA damage.108
Cocaine-induced heart failure management includes cessation, guideline-directed medical therapy, and, in severe cases, cardiac transplantation.109 Selenium supplementation has been shown to improve cardiac function and prevent dysfunction induced by cocaine in rats, indicating oxidative stress’s significant role in cocaine-induced cardiotoxicity.110
In pronounced cases of drug-induced cardiotoxicity, presenting as cardiovascular shock or cardiac arrest, circulatory assistance, such as extracorporeal life support (ECLS), may be critical. However, ECLS can be associated with complications like limb ischemia, hemorrhage, and embolism.111
Conclusion
Cardiotoxicity caused by drugs presents a significant challenge in the development and use of various medications and has been a primary concern in drug safety. Therefore, an in-depth understanding of the pathological mechanisms behind drug-induced cardiotoxicity of certain drugs is crucial. This knowledge aids in future research for the development of cardioprotective agents and in reducing the morbidity and mortality associated with drug-induced cardiotoxicity. This review has presented an extensive discussion on the drug-receptor mechanisms underlying drug-induced cardiotoxicity, emphasizing that receptors are key factors in any pathological mechanism. The mechanisms of drug-induced cardiotoxicity and their management discussed herein represent significant and prospective areas for future drug development, safety pharmacology, and pharmacovigilance studies.
Acknowledgement
We extend our sincere gratitude to all the researchers and scientists whose work has contributed to our understanding of the pathologic mechanisms of cardiotoxic drugs. We also wish to thank our colleagues and peers for their constructive feedback and support throughout the writing process.
Conflicting Interest
None
Funding Information
Not Applicable
References
- Haddad LM, Winchester JF, Shannon MW, Borron SW, Burns M. Haddad and Winchester’s Clinical Management of Poisoning and Drug Overdose. Saunders/Elsevier; 2007.
- Bryson PD. Comprehensive Review in Toxicology for Emergency Clinicians. Taylor & Francis; 1996.
- Rosano TG. Ellenhorn’s Medical Toxicology: Diagnosis and Treatment of Human Poisoning, 2nd ed. Matthew J. Ellenhorn, Seth Schonwald, Gary Ordog, and Jonathan Wasserberger. Baltimore, MD: Williams and Wilkins, 1997, 2047 pp., $199, ISBN 0–683-30031–8. Clinical Chemistry. 1998;44(2):366-366.
CrossRef - Brunton L, Chabner B, Knollman B. Goodman and Gilman’s the Pharmacological Basis of Therapeutics, Twelfth Edition. McGraw Hill Professional; 2011.
- Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments. Nature Reviews Cardiology. 2015;12(9):547-558.
CrossRef - Mamoshina P, Rodriguez B, Bueno-Orovio A. Toward a broader view of mechanisms of drug cardiotoxicity. Cell Reports Medicine. 2021;2(3):100216.
CrossRef - Pistillucci G, Ciorra AA, Sciacca V, Raponi M, Rossi R, Veltri E. Troponina I e Peptide NatriureticoCerebrale (BNP) come biomarcatoripredittivi di cardiotossicitànellepazientiaffette da carcinoma dellamammella in terapiaadiuvante con antracicline e trastuzumab [Troponin I and B-type Natriuretic Peptide (BNP) as biomarkers for the prediction of cardiotoxicity in patients with breast cancer treated with adjuvant anthracyclines and trastuzumab]. Clin Ter. 2015;166(1):e67-e71.
- Varga ZV, Ferdinandy P, Liaudet L, Pacher P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol Heart Circ Physiol. 2015;309(9):H1453-H1467.
CrossRef - Bloom MW, Hamo CE, Cardinale D, et al. Cancer Therapy-Related Cardiac Dysfunction and Heart Failure: Part 1: Definitions, Pathophysiology, Risk Factors, and Imaging. Circ Heart Fail. 2016;9(1):e002661.
CrossRef - Hiraoka M. Mechanisms of Drug-Induced Cardiac Toxicity. Electrical Diseases of the Heart.:691-704.
CrossRef - Kim S, Park T. Acute and Chronic Effects of Cocaine on Cardiovascular Health. International Journal of Molecular Sciences. 2019;20(3):584.
CrossRef - Burns, L. World drug report 2013 by United Nations Office on Drugs and Crime New York: United Nations, 2013 ISBN: 978-921-056168-6, 151 pp. Grey literature. Drug Alcohol Rev. 2014;33:216.
CrossRef - Silva MO, Roth D, Reddy KR, Fernandez JA, Albores-Saavedra J, Schiff ER. Hepatic dysfunction accompanying acute cocaine intoxication. J Hepatol. 1991;12(3):312-315.
CrossRef - Degenhardt L, Singleton J, Calabria B, et al. Mortality among cocaine users: A systematic review of cohort studies. Drug and Alcohol Dependence. 2011;113(2):88-95.
CrossRef - Tortajada S, Herrero MJ, Domingo-Salvany A, et al. Psychiatric morbidity among cocaine and heroin users in the community.Adicciones. 2012;24(3):201-210.
CrossRef - Qureshi AI, Chaudhry SA, Suri MF. Cocaine use and the likelihood of cardiovascular and all-cause mortality: data from the Third National Health and Nutrition Examination Survey Mortality Follow-up Study. J VascInterv Neurol. 2014;7(1):76-82.
- Schwartz BG, Rezkalla S, Kloner RA. Cardiovascular effects of cocaine. Circulation. 2010;122(24):2558-2569.
CrossRef - Lange RA, Hillis LD. Cardiovascular complications of cocaine use. N Engl J Med. 2001;345(5):351-358.
CrossRef - Kloner RA, Rezkalla SH. Cocaine and the heart. N Engl J Med. 2003;348(6):487-488.
CrossRef - Bourque SL, Davidge ST, Adams MA. The interaction between endothelin-1 and nitric oxide in the vasculature: new perspectives. Am J PhysiolRegulIntegr Comp Physiol. 2011;300(6):R1288-R1295.
CrossRef - Nemeth BT, Varga ZV, Wu WJ, Pacher P. Trastuzumab cardiotoxicity: from clinical trials to experimental studies. Br J Pharmacol. 2017;174(21):3727-3748.
CrossRef - Lee KF, Simon H, Chen H, Bates B, Hung MC, Hauser C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. 1995;378(6555):394-398.
CrossRef - Strasser F, Betticher DC, Suter TM. Trastuzumab and breast cancer. N Engl J Med. 2001;345(13):996.
CrossRef - Sawyer DB, Zuppinger C, Miller TA, Eppenberger HM, Suter TM. Modulation of anthracycline-induced myofibrillar disarray in rat ventricular myocytes by neuregulin-1beta and anti-erbB2: potential mechanism for trastuzumab-induced cardiotoxicity. 2002;105(13):1551-1554.
CrossRef - Holbro, T., and Hynes, N. E. ErbB receptors: directing key signaling networks throughout life. Rev. Pharmacol. Toxicol. 2004; 44:195–217.
CrossRef - Grazette, L. P., Boecker, W., Matsui, T., Semigran, M., Force, T. L., Hajjar, R. J., et al. Inhibition of ErbB2 causes mitochondrial dysfunction in cardiomyocytes: implications for herceptin-induced cardiomyopathy. Am. Coll. Cardiol.2004;44:2231–2238.
CrossRef - Fuchs IB, Landt S, Bueler H, et al. Analysis of HER2 and HER4 in human myocardium to clarify the cardiotoxicity of trastuzumab (Herceptin). Breast Cancer Res Treat. 2003;82(1):23-28.
CrossRef - Szymanski MW, Singh DP. Isoproterenol. Nih.gov. Published July 14, 2019. https://www.ncbi.nlm.nih.gov/books/NBK526042/
- Rona G. Catecholamine cardiotoxicity. J Mol Cell Cardiol. 1985;17(4):291-306.
CrossRef - Wexler BC, Greenberg BP. Protective effects of clofibrate on isoproterenol-induced myocardial infarction in arteriosclerotic and non-arteriosclerotic rats. Atherosclerosis. 1978;29(3):373-395.
CrossRef - Van Vleet, J. F., Ferrans, J. V., and Herman, E. Cardiovascular and skeletal muscle system. In Handbook of Toxicologic Pathology (W. M. Haschek,C. G. Rousseaux, and M. A. Wallig, eds). 2002;2:363–455.
CrossRef - Boutet, M., Huttner, I., and Rona, G. Alteration of the sarcolemmal membrane in catecholamine-induced cardiac muscle cell injury: In vivo studies with fine structural diffusion tracer horseradish peroxidase. Lab Invest.1976;34:482–88.
- Filipský T, Zatloukalová L, Mladěnka P, Hrdina R. Acute initial haemodynamic changes in a rat isoprenaline model of cardiotoxicity. Hum Exp Toxicol. 2012;31(8):830-843.
CrossRef - Blasig IE, Zipper J, Muschick P, Modersohn D, Löwe H. Absolute and relative myocardial ischemia by isoproterenol overdosage. Biomed Biochim Acta. 1985;44(11-12):1641-1649.
- Dhalla NS, Dent MR, Arneja AS. Pathogenesis of catecholamine-induced cardiomyopathy. In: Acosta D Jr, ed. Cardiovascular Toxicology. Vol. 25 (Target Organ Toxicology Series, Hayes AW, Thomas JA, Gardner DE, eds). 4th ed. New York: Informa HealthCare; 2008:207-262.
- Azuma J, Hamaguchi T, Ohta H, et al. Calcium overload-induced myocardial damage caused by isoproterenol and by adriamycin: Possible role of taurinein its prevention. Adv ExpMedBiol.1987;217:167–179.
CrossRef - Tappia PS, Hata T, Hozaima L, Sandhu MS, Panagia V, Dhalla NS. Role of oxidative stress in catecholamine-induced changes in cardiac sarcolemmal Ca2+ transport. Arch BiochemBiophys. 2001;387(1):85-92.
CrossRef - Nichols CB, Rossow CF, Navedo MF, et al. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ Res. 2010;107(6):747-756.
CrossRef - Pinelli A, Trivulzio S, Tomasoni L, Brenna S, Bonacina E, Accinni R. Isoproterenol-induced myocardial infarction in rabbits. Protection by propranolol or labetalol: a proposed non-invasive procedure. Eur J Pharm Sci. 2004;23(3):277-285.
CrossRef - von Känel R, Mills PJ, Ziegler MG, Dimsdale JE. Effect of beta2-adrenergic receptor functioning and increased norepinephrine on the hypercoagulable state with mental stress. Am Heart J. 2002;144(1):68-72.
CrossRef - Pinelli A, Trivulzio S, Tomasoni L, et al. Myocardial infarction non-invasively induced in rabbits by administering isoproterenol and vasopressin: protective effects exerted by verapamil. Fundam Clin Pharmacol. 2004;18(6):657-667.
CrossRef - Remião F, Carmo H, Carvalho F, Bastos ML. The study of oxidative stress in freshly isolated Ca(2+)-tolerant cardiomyocytes from the adult rat. Toxicol In Vitro. 2001;15(4-5):283-287.
CrossRef - Remião F, Carvalho M, Carmo H, Carvalho F, Bastos ML. Cu2+-induced isoproterenol oxidation into isoprenochrome in adult rat calcium-tolerant cardiomyocytes. Chem Res Toxicol. 2002;15(6):861-869.
CrossRef - Hašková P, Koubková L, Vávrová A, et al. Comparison of various iron chelators used in clinical practice as protecting agents against catecholamine-induced oxidative injury and cardiotoxicity. 2011;289(2-3):122-131.
CrossRef - Hašková P, Kovaříková P, Koubková L, Vávrová A, Macková E, Simůnek T. Iron chelation with salicylaldehyde isonicotinoyl hydrazone protects against catecholamine autoxidation and cardiotoxicity. Free Radic Biol Med. 2011;50(4):537-549.
CrossRef - Bindoli A, Rigobello MP, Deeble DJ. Biochemical and toxicological properties of the oxidation products of catecholamines. Free Radic Biol Med. 1992;13(4):391-405.
CrossRef - Zatloukalová L, Filipský T, Mladěnka P, et al. Dexrazoxane provided moderate protection in a catecholamine model of severe cardiotoxicity. Can J PhysiolPharmacol. 2012;90(4):473-484..
CrossRef - Mladĕnka P, Kalinowski DS, Haskova P, et al. The novel iron chelator, 2-pyridylcarboxaldehyde 2-thiophenecarboxyl hydrazone, reduces catecholamine-mediated myocardial toxicity. Chem Res Toxicol. 2009;22(1):208-217.
CrossRef - Mladenka P, Semecký V, Bobrovová Z, et al. The effects of lactoferrin in a rat model of catecholamine cardiotoxicity. 2009;22(2):353-361. doi:10.1007/s10534-008-9172-5.
CrossRef - Waugh J, Keating GM, Plosker GL, Easthope S, Robinson DM. Pioglitazone. Drugs. 2006;66(1):85-109.
CrossRef - Kalra S, Shukla R. Pioglitazone: Indian perspective. Indian Journal of Endocrinology and Metabolism. 2011;15(4):294.
CrossRef - Gale EA. Lessons from the glitazones: a story of drug development. Lancet. 2001;357(9271):1870-1875.
CrossRef - Baraka, A., and Abdelgawad, H. (2010). Targeting apoptosis in the heart of streptozotocin-induced diabetic rats. Cardiovasc. Pharmacol. Ther. 15, 175– 181.
CrossRef - Ma W, Wei S, Zhang B, Li W. Molecular Mechanisms of Cardiomyocyte Death in Drug-Induced Cardiotoxicity. Frontiers in Cell and Developmental Biology.2020;8:34.
CrossRef - Petrovan RJ, Kaplan CD, Reisfeld RA, Curtiss LK. DNA vaccination against VEGF receptor 2 reduces atherosclerosis in LDL receptor-deficient mice. ArteriosclerThrombVasc Biol. 2007;27(5):1095-1100.
CrossRef - Bhardwaj S, Roy H, Babu M, Shibuya M, Yla-Herttuala S. Adventitial gene transfer of VEGFR-2 specific VEGF-E chimera induces MCP-1 expression in vascular smooth muscle cells and enhances neointimal formation. 2011;219(1):84-91.
CrossRef - Lohela M, Bry M, Tammela T, Alitalo K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. CurrOpin Cell Biol. 2009;21(2):154-165.
CrossRef - Sarabipour S, Ballmer-Hofer K, Hristova K. VEGFR-2 conformational switch in response to ligand binding. 2016;5:e13876.
CrossRef - Masuda T, Muto S, Fujisawa G, et al. Heart angiotensin II-induced cardiomyocyte hypertrophy suppresses coronary angiogenesis and progresses diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2012;302(9):H1871-H1883.
CrossRef - Zheng L, Han P, Liu J, et al. Role of copper in regression of cardiac hypertrophy. Pharmacol Ther.2015;148:66-84.
CrossRef - Song HK, Kim J, Lee JS, et al. Pik3ip1 modulates cardiac hypertrophy by inhibiting PI3K pathway.PLoS One. 2015;10(3):e0122251. Published 2015 Mar 31.
CrossRef - Journey JD, Bentley TP. Theophylline Toxicity. Published 2020. https://www.ncbi.nlm.nih.gov/ books/NBK532962/
- Higbee MD, Kumar M, Galant SP. Stimulation of endogenous catecholamine release by theophylline: a proposed additional mechanism of action for theophylline effects. J Allergy Clin Immunol. 1982;70(5):377-382.
CrossRef - Harbin AD, Gerson MC, O’Connell JB. Simulation of acute myopericarditis by constrictive pericardial disease with endomyocardial fibrosis due to methysergide therapy. J Am Coll Cardiol. 1984;4(1):196-199.
CrossRef - Mason JW, Billingham ME, Friedman JP. Methysergide-induced heart disease: a case of multivalvular and myocardial fibrosis. 1977;56(5):889-890.
CrossRef - Redfield MM, Nicholson WJ, Edwards WD, Tajik AJ. Valve disease associated with ergot alkaloid use: echocardiographic and pathologic correlations. Ann Intern Med. 1992;117(1):50-52.
CrossRef - Tan C, Tasaka H, Yu KP, Murphy ML, Karnofsky DA. Daunomycin, an antitumor antibiotic, in the treatment of neoplastic disease. Clinical evaluation with special reference to childhood leukemia. 1967;20(3):333-353.
CrossRef - Ewer MS, Ewer SM. Troponin I Provides Insight Into Cardiotoxicity and the Anthracycline-Trastuzumab Interaction. Journal of Clinical Oncology. 2010;28(25):3901-3904.
CrossRef - Scott JM, Khakoo A, Mackey JR, Haykowsky MJ, Douglas PS, Jones LW. Modulation of Anthracycline-Induced Cardiotoxicity by Aerobic Exercise in Breast Cancer. Circulation. 2011;124(5):642-650.
CrossRef - Ma Y, Zhang X, Bao H, et al. Correction: Toll-Like Receptor (TLR) 2 and TLR4 Differentially Regulate Doxorubicin Induced Cardiomyopathy in Mice. Peng T, ed. PLoS ONE. 2012;7(10).
CrossRef - Yao Y, Xu X, Zhang G, Zhang Y, Qian W, Tao R. Role of HMGB1 in doxorubicin-induced myocardial apoptosis and its regulation pathway. Basic Research in Cardiology. 2012;107(3).
CrossRef - Ichikawa Y, Ghanefar M, Bayeva M, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. Journal of Clinical Investigation. 2014;124(2):617-630.
CrossRef - Zhang T, Zhang Y, Cui M, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress–induced myocardial necroptosis. Nature Medicine. 2016;22(2):175-182.
CrossRef - Shiga T, Hiraide M. Cardiotoxicities of 5-Fluorouracil and Other Fluoropyrimidines. Current Treatment Options in Oncology. 2020;21(4).
CrossRef - Madeddu C, Deidda M, Piras A, et al. Pathophysiology of cardiotoxicity induced by nonanthracycline chemotherapy. Journal of Cardiovascular Medicine. 2016;17:e12-e18.
CrossRef - Focaccetti C, Bruno A, Magnani E, et al. Effects of 5-Fluorouracil on Morphology, Cell Cycle, Proliferation, Apoptosis, Autophagy and ROS Production in Endothelial Cells and Cardiomyocytes. Wang YJ, ed. PLOS ONE. 2015;10(2):e0115686.
CrossRef - Cianci G, Morelli MC, Cannita K, et al. Prophylactic options in patients with 5-fluorouracil-associated cardiotoxicity. British Journal of Cancer. 2003;88(10):1507-1509.
CrossRef - Shoemaker L, Arora UK, Rocha MS. 5-Fluorouracil–Induced Coronary Vasospasm. Cancer Control. 2004;11(2):46-49.
CrossRef - Lamberti M, Porto S, Zappavigna S, et al. A mechanistic study on the cardiotoxicity of 5-fluorouracil in vitro and clinical and occupational perspectives. Toxicology Letters. 2014;227(3):151-156.
CrossRef - Kalam K, Marwick TH. Role of cardioprotective therapy for prevention of cardiotoxicity with chemotherapy: A systematic review and meta-analysis. European Journal of Cancer. 2013;49(13):2900-2909.
CrossRef - Altieri P, Murialdo R, Barisione C, et al. 5-fluorouracil causes endothelial cell senescence: potential protective role of glucagon-like peptide 1. British Journal of Pharmacology. 2017;174(21):3713-3726.
CrossRef - Alter P, Herzum M, Soufi M, Schaefer J, Maisch B. Cardiotoxicity of 5-Fluorouracil. Cardiovascular & Hematological Agents in Medicinal Chemistry. 2006;4(1):1-5.
CrossRef - Sorrentino MF, Kim J, Foderaro AE, Truesdell AG. 5-fluorouracil induced cardiotoxicity: Review of the literature. Cardiology Journal. 2012;19(5):453-457.
CrossRef - Haugnes HS, Wethal T, Aass N, et al. Cardiovascular Risk Factors and Morbidity in Long-Term Survivors of Testicular Cancer: A 20-Year Follow-Up Study. Journal of Clinical Oncology. 2010;28(30):4649-4657.
CrossRef - Ma H, Jones KR, Guo R, Xu P, Shen Y, Ren J. Cisplatin compromises myocardial contractile function and mitochondrial ultrastructure: Role of endoplasmic reticulum stress. Clinical and Experimental Pharmacology and Physiology. 2010;37(4):460-465.
CrossRef - McGettigan P, Henry D. Use of Non-Steroidal Anti-Inflammatory Drugs That Elevate Cardiovascular Risk: An Examination of Sales and Essential Medicines Lists in Low-, Middle-, and High-Income Countries. Turnbull FM, ed. PLoS Medicine. 2013;10(2):e1001388.
CrossRef - Egan K, Wang M, Lucitt MB, et al. Cyclooxygenases, Thromboxane, and Atherosclerosis. Circulation. 2005;111(3):334-342.
CrossRef - Rabausch K, Bretschneider E, Sarbia M, et al. Regulation of Thrombomodulin Expression in Human Vascular Smooth Muscle Cells by COX-2–Derived Prostaglandins. Circulation Research. 2005;96(1).
CrossRef - Lin Y, Bai L, Chen W, Xu S. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert opinion on therapeutic targets. 2010;14(1):45-55.
CrossRef - Lin Y., Bai L., Chen W., Xu S. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opinion on Therapeutic Targets. 2010;14(1):45–55.
CrossRef - Il Yeob Kim, Su Young Park, You Ra Kang, Thapa D, Han Gon Choi, Jung Ae Kim. Role of nonsteroidal anti-inflammatory drug-activated gene-1 in docetaxel-induced cell death of human colorectal cancer cells with different p53 status. Archives of Pharmacal Research. 2011;34(2):323-330.
CrossRef - Zhang Y, Dai Q, Wu S, et al. Susceptibility for NSAIDs-Induced Apoptosis Correlates to p53 Gene Status in Gastric Cancer Cells. Cancer Investigation. 2008;26(9):868-877.
CrossRef - Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochemical Pharmacology. 2006;71(10):1397-1421.
CrossRef - Singh BK, Pillai KK, Haque SE, Dubey K. Diclofenac sodium, a nonselective nonsteroidal antiinflammatory drug aggravates doxorubicin-induced cardiomyopathy in rats. J Cardiovasc Pharmacol. 2010;55:139-44.
CrossRef - Cocco T, Di M, Papa P, Lorusso M. Arachidonic acid interaction with the mitochondrial electron transport chain promotes reactive oxygen species generation. Free Radical Biology and Medicine. 1999;27(1-2):51-59.
CrossRef - Bistrovic P, Lucijanic M. Remdesivir might induce changes in electrocardiogram beyond bradycardia in patients with coronavirus disease 2019—The pilot study. Journal of Medical Virology. 2021;93(10):5724-5725.
CrossRef - Parham WA, Mehdirad AA, Biermann KM, Fredman CS. Journal of Interventional Cardiac Electrophysiology. 2001;5(1):71-74.
CrossRef - Cueva J, Antolín S, Calvo L, Fernández I, Ramos M, de Paz L, et al. Galician consensus on management of cardiotoxicity in breast cancer: risk factors, prevention, and early intervention. Clinical and Translational Oncology. 2017:1–12.
CrossRef - WRITING COMMITTEE MEMBERS, Yancy CW, Jessup M, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. 2013;128(16):e240-e327.
- Mukku RB, Fonarow GC, Watson KE, Ajijola OA, Depasquale EC, Nsair A, et al. Heart Failure Therapies for End-Stage Chemotherapy–Induced Cardiomyopathy. Journal of cardiac failure. 2016;22(6):439–48.
CrossRef - Bianco CM, Al-Kindi SG, Oliveira GH. Advanced Heart Failure Therapies for Cancer Therapeutics–Related Cardiac Dysfunction. Heart Failure Clinics. 2017;13(2):327–36.
CrossRef - Varricchi G et al. Antineoplastic drug-induced cardiotoxicity: A redox perspective. Frontiers in Physiology. 2018;9:167.
CrossRef - McGowan JV et al. Anthracycline chemotherapy and cardiotoxicity. Cardiovascular Drugs and Therapy. 2017;31(1):63-75.
CrossRef - Ichihara S, Yamada Y, Kawai Y, et al. Roles of oxidative stress and Akt signaling in doxorubicin cardiotoxicity. Biochemical and Biophysical Research Communications. 2007;359(1):27-33.
CrossRef - Schimmel KJM, Richel DJ, van den Brink RBA, Guchelaar HJ. Cardiotoxicity of cytotoxic drugs. Cancer Treatment Reviews. 2004;30(2):181-191.
CrossRef - Cardinale D, Iacopo F, Cipolla CM. Cardiotoxicity of Anthracyclines. Frontiers in Cardiovascular Medicine. 2020;7.
CrossRef - Lyu YL, Kerrigan JE, Lin CP ., et al. Topoisomerase II Mediated DNA Double-Strand Breaks: Implications in Doxorubicin Cardiotoxicity and Prevention by Dexrazoxane. Cancer Research. 2007;67(18):8839-8846.
CrossRef - Elkattawy S, Alyacoub R, Al-Nassarei A, Younes I, Ayad S, Habib M. Cocaine induced heart failure: report and literature review. Journal of community hospital internal medicine perspectives. 2021;11(4):547-550.
CrossRef - Moritz F, Monteil C, Isabelle M, et al. Selenium diet-supplementation improves cocaine-induced myocardial oxidative stress and prevents cardiac dysfunction in rats. Fundamental and Clinical Pharmacology. 2004;18(4):431-436.
CrossRef - Ashrafian H, Athanasiou T. Extracorporeal life support for severe drug-induced cardiotoxicity: a promising therapeutic choice. Critical Care. 2009;13(5):187.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.



