

How to Cite | Publication History | PlumX Article Matrix
The Emergence of In-Silico Models in Drug Target Interaction System: A Comprehensive Review
Janet Reshma Jeyasingh1 and Glory Josephine I2*
and Glory Josephine I2*
1Department of Computer Science, University of Massachusetts, Amherst, Massachusetts, United States.
2Department of Pharmacology, Sree Balaji Medical College and Hospital, Bharath Institute of Higher Education and Research (BIHER), Chennai, India.
corresponding Author E-mail:dr.gloryj@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3198

ABSTRACT: The dawn of computational models in healthcare has revolutionised the drug development industry. The wet lab experiments entail enormously expensive and laborious procedures. As a result, the applications of computational designs have been a better replacement for manual experimentations. Identifying drug-target interaction (DTI) is a vital drug design process. In this review, we have explored the various computational methodologies actively used in the field of DTI prediction. We have hierarchically categorised the models into three broad domains: ligand-based, structure-based and chemogenic. We have further classified the domains into their subcategories. The functioning and latest developments achieved in each subcategory are further analysed in depth. This review offers a comprehensive overview of the tools and methodologies of each model. We have also compared the advantages and limitations of each model in every category. Finally, we look into the future scope of the machine learning models by addressing the possible difficulties faced in DTI. This article serves as an insight into the various models used in DTI prediction.
KEYWORDS: Drug-target interaction (DTI); Chemogenic models; Ligand-based; Structure-based
Download this article as:| Copy the following to cite this article: Jeyasingh J. R, Josephine I. G. The Emergence of In-Silico Models in Drug Target Interaction System: A Comprehensive Review. Biotech Res Asia 2024;21(1). |
| Copy the following to cite this URL: Jeyasingh J. R, Josephine I. G. The Emergence of In-Silico Models in Drug Target Interaction System: A Comprehensive Review. Biotech Res Asia 2024;21(1). Available from: https://bit.ly/3xaS609 |
Introduction
The increasing incidence of the disease burden necessitates the development of newer molecular entities. The average cost of developing a new drug range from 1 million to 5 billion 1,2. Many challenges, such as patient heterogeneity, lack of targets and biomarkers, lack of valid animal models, regulatory issues, cost, and prolonged drug development stages are several significant hindrances faced in the drug research and development (R&D) field 3. The various stages of preclinical drug development are target identification, validation, hit identification and lead prediction to optimisation. High throughput screening and in-vivo & and vitro screening methods have helped to discover the molecules in the initial stages accounting from target identification to lead prediction through various biological assays 4-6. However, the expensive and time-consuming initial processes entail newer models for lead identification. Computer-aided drug design technologies (CADD) limit animal use and are cost-effective in identifying suitable drug candidates 7. Furthermore, In-silico methods collaborate with in-vitro methods in selecting the lead component for optimisation. These innovative technologies help in drug designing/discovery processes by examining the drug-target interactions (DTI) and the binding affinities (DTBA). The technical advancement and development of computational methods with informative bioresources could reduce the significant burden of drug development.
The drug-target interaction plays a vital role in drug discovery and drug repurposing. The polypharmacological properties of a drug and the ability to bind with multiple targets favour drug repurposing. Novel computational polypharmacology approaches have been developed for designing multi-target agents 8. Identifying and validating the drug target are the initial phases of drug discovery. The interaction of an ion channel, enzymes, G protein-coupled receptor, and nuclear receptor modulates target protein effects. Drug-target prediction helps to understand the mechanism, therapeutic effect, and side effects of drugs. Several factors affect the association of the drug target interaction and experimental validation, making it a cumbersome process. Nowadays, multi-target drug development is imperative to increase drug efficacy and overcome resistance 9,10,11. Considering these facts, we need appropriate computational models to detect the novel potential drug target association. Cheminformatics, data-mining techniques, and structural databases are used to identify the target site prediction. The possible target identification can be done by comparing these HCS hit structures with 3D shapes of annotated compounds. Once lead identification is done, lead optimisation can proceed with computer designing.
Computational methods are applied to various phases of the drug development procedure and produce fruitful outcomes. Drug target interaction is one of the crucial steps in drug development. Ligand-based drug design, structure-based drug design and chemogenic approach are the three main classes that constitute in silico methods. The former two classes are a part of CADD tools. CADD is a well-renowned computer tool that generates graphical simulations of a compound. CADD involves the following principles: (i) Utilisation of potential to rationalise drug discovery and design. (ii) Requires information about ligands and targets for identifying and repurposing the drugs. (iii) Designing filters that remove unwanted molecules and perform eligible candidate selection. Ligand-based CADD adheres to the idea that similar molecules and proteins bind together. On the other hand, structure-based CADD procures information from the three-dimensional structures of targets to streamline their binding with drugs. This CADD includes three different groups: molecular docking, virtual molecular screening, and molecular dynamics. Among these, molecular docking is widely used for simulation purposes. Some popular molecular docking tools are GOLD, AutoDock v4, AutoDock Vina, FlexX, and Dock.
Chemogenic approaches overcome the limitations of the above-stated methods. These approaches include machine learning and deep learning methods that forecast the interactions. They consider chemogenic and biological information as target sources for an interaction. They combine all the predictions made for interaction from all the different sources to provide the outcome. Feature-based and similarity-based are two types of categories in chemogenic approaches. Feature-based approaches obtain the features from known interaction sources, whereas similarity-based techniques obtain input from the neighbour nodes’ features.
Virtual screening and de novo drug designing methods synthesise suitable drug candidates based on the binding site of the target protein with greater affinity 12. Artificial intelligence (Machine learning and deep learning) integrates different computational pharmacology methods to improvise drug screening and designing processes 13, 14. Furthermore, these effective algorithms could be an ideal practical tool in various drug development stages. The multiscale de novo drug design, which combines the QSAR models, can effectively improve the properties of the drug and favours personalised medicine 15.
In this review, we have discussed the various strategies that forecast drug target complexes. We have also discussed the subcategories involved in each strategy and analysed their merits and demerits.
Rise of Artificial Intelligence in DTI
The advent of computational biology has marked a significant breakthrough in computation strategies like relative binding energies, active core matter and lead optimization 16-18. Further, the insurgence of computer-based models, ranging from simple software tools to complex DL models, is gaining popularity in the field of drug design. The computational methodologies are again widely classified into three main broad categories: Ligand-based, structure-based and chemogenic approaches. One such method that has gained popularity in drug design research is Computer-aided drug design. CADD are computational tools used to quantify and modify characteristics of possible ligands to expedite the drug design process. Ligand-based and Structure-based are two different CADD methods that are classified based on the availability and structural features of the compounds.
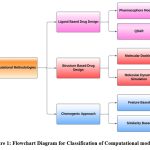 |
Figure 1: Flowchart Diagram for Classification of Computational models Click here to view Figure |
Ligand – Drug Molecular Binding
Ligand-based approach predicts binding possibilities based on similarities in the physiological properties between a drug-protein and target. This method is also referred to as the indirect protocol in pharmaceutical research, as it relies on the structural data of the known ligand molecules to understand the structural properties of ligands that share the same pharmacological behaviour 19-21. Ligand-based models comprise three models: Pharmacophore Modelling and Quantitative Structure-Activity Relationship (QSAR). The former two are widely used and are discussed in this section.
The first approach, the pharmacophore model, gives a description of the minimum physiological characteristics a molecule must possess to bind to the target. The pharmacophore query encodes the interaction pattern in a three-dimensional way. Different pharmacophore queries are structured for identification purposes based on the following cases. (i) If the structure of the target is present, then the model is constructed by analysing the action mode of the receptor and a drug molecule involved. (ii) If the structure of the protein is not known and only the active molecule, then pharmacophore fingerprints can be applied to get similar molecules. A pharmacophore fingerprint is a string which describes the frequency of every possible combination of molecular interaction features in the string 22. One of the popular pharmacophore modelling software is LigandScout. It can construct queries automatically from a protein database which contains interaction files. Some of the other programs are DISCO, GASP, RAPID and HypoGen. Pharmacophore modelling has several demerits: (i) Difficult to construct as the number of ligands and their flexibilities increase. (ii) lacks good scoring function (iii) Highly dependent on priorly available conformation database. These reasons make pharmacophore a non-ideal prediction method.
The second one, QSAR, is a mathematical model that finds and recapitulates the relationships between trends in structural changes and biological endpoints to discover which chemical properties determine their biological activities. In drug discovery, QSAR identifies structures that could inhibit effects on specific targets and are less toxic (non-specific activity) 23. Three-dimensional structural knowledge was introduced in QSAR, resulting in the formation of 3D-QSAR. This QSAR captures the interaction between drug and target molecules in 3D format and reflects the changes in their energy and pattern. Thus, studying the structure-activity relationship 24.
One of the major bottlenecks in this drug design model is that they cannot work when both the target and interacting protein structures are unknown. Another drawback is that ligand-based methods perform poorly when the number of known ligands is insufficient 25. The high computational cost involved in these methods surpasses their usage. QSAR has an essential role in the drug design process as they are cheap compared to the medium throughput in vitro and low throughput. These models evolved to support 3D structural data. However, the advent of SB-CADD resulted in the replacement of this drug design. The more computational power and accuracy given by SBDD makes it a preferred CADD. The famous ligand-based tools are given in Table 1.
Table 1: Ligand based drug design – Pharmacophore modelling Software Techniques
| ALGORITHM USED | SOFTWARE/ PROGRAMS | DESCRIPTION |
| Pharmacophore Modelling | PharmaGist | A web server for pharmacophore detection which requires a set of structures of drugs as input that are known to bind to the target. The pharmacophores are constructed by multiple flexible alignments of the input. |
| Pharmacophore Modelling | LigandScout | A fully integrated platform which is used for accurate virtual screening. They develop 3D chemical feature pharmacophore models |
| Pharmacophore Modelling | MOE | Pharmacophore discovery application used for fragment-based, ligand and structure-based design projects. Searches for novel active compounds when no receptor is available |
| Pharmacophore Modelling | Catalyst | Does 3D database building and searching; ligand conformer generation and analysis tools. Part of Discovery Studio |
| QSAR | Cloud 3D QSAR | A web tool that integrates the functions of molecular structure generation, alignment, and molecular interaction field (MIF) computing. Final analysis to provide one solution. |
Structure Based Computer Aided Drug Design
Structure-based computer-aided drug design (SBDD) is based on the knowledge of target structure to compute minimal binding free energy to get ligand-receptor complex. This type helps design entities that require minimal energy and hold onto the target 26. They are used to screen a macromolecule binding to the ligand location virtually. However, this model requires a huge collection of experimentally solved to achieve precision. Molecular Docking and Molecular dynamics simulation are the most prevalent SBDD models.
Molecular Docking simulations
One of the popular methods in this CADD is molecular docking. This methodology involves graphically simulating the bonding between two molecules by computing the binding affinity and thus identifying a possible drug target. Docking methods are used to get the potential ligands and can estimate the binding parameters of the complex in prior. Most docking tools deploy known stochastic search algorithms, namely, Evolutionary algorithms, Monte Carlo algorithms and fragment-based algorithms. These search algorithms can follow two kinds of docking simulations. When the mobility of both receptor and ligand are static, it is rigid docking. The other type is flexible docking, in which both compounds are mobile and flexible. The binding energy and size are computed, and the pose is finalized 26. Tools like DOCK, FLOG, GOLD, FlexX and ICM that utilise these algorithms offer high throughput.
Semi-flexible docking using Monte Carlo algorithms
The Monte Carlo algorithm is a semi-flexible docking method applied to the probe’s degrees of freedom. The algorithm utilises a conformational search strategy to generate different ligand positions by considering properties like affinity, bond rotation, compound translation or rotation. Energy-based selection criteria are used to evaluate the generated complex. If it passes the criterion, the entity is used to generate the next conformation. Most of them use the Metropolis criterion for this. Metropolis criterion generally accepts steps that lower the total energy and rarely takes steps that increase energy to avoid being stuck in the local energy minimum. The tools which use this algorithm are MCDock, AutoDock, ICM, QXP and Affinity 27.
Predicting docking positions with full ligand flexibility
One of the renowned MC methods, MCDock tool, a computer program that automatically predicts a ligand’s binding mode by allowing full flexibility of ligands. The scoring function used in this algorithm is a combination of interaction and conformational energies 28. Primarily, a ligand is placed randomly into the target, considering both are in a rigid state. The non-clashing poses are identified by the scoring function to evaluate them based on the minimal contacts made. Later, Metropolis sampling is done to sample the binding spots.
Blind Docking mechanism using protein structures
The latest development of this simulation is EDock. EDock focuses on predicting precise blind docking by feeding on low-resolution predicted protein structures. This model comprises five sequential steps: ligand-binding site prediction, binding pocket construction, initial docking pose generation, REMC docking simulation, and final model selection. In the first step, the final prediction is chosen by combining results from algorithms like S-SITE through a linear SVM consensus model. The binding pocket is then obtained through negative imaging which is used by a modified graph-matching algorithm to generate the initial ligand docking conformations 29 where every node represents a pair of ligand atoms and the binding pocket. The penultimate step, REMC (Replica Monte Carlo simulation) is used to improve the efficiency of sampling of the docking system and involves rigid-body ligand translations and rotations. Scoring techniques like XSCORE and SPICKER methods are used to get the best performance with both predicted and experimental receptor structures 30.
Many of the docking algorithms require the user to specify the binding site prior. Sometimes, the native binding pocket may be oblivious to the end users. The Monte Carlo method, EDock, overcomes this limitation by supporting blind docking. Despite MC models’ success in accurate prediction, they bear high computational costs to long model timescales on protein-ligand binding and unbinding. Since these methods produce different orientations of a drug molecule, these models can be used only when the 3D structure of compounds is available. Moreover, they are difficult to apply to member proteins as their structures are very complex.
Evolutionary Semi Flexible Stochastic Algorithm
Another example of semi-flexible stochastic algorithms is evolutionary algorithms. These algorithms are generally iterative stochastic optimisation procedures similar to the evolution process. A genetic algorithm identifies possible solutions to combinatorial optimisation problems for search problems where the ligands and proteins are arranged like that of the DNA sequence. This representation is called a state variable. The fitness of a newly formed complex is the total interaction energy between the molecules in the complex. A complex scoring function, incorporating factors like mutation, crossover rates and several evolutionary rounds, is used to calculate this fitness 31-33. The methodology of the genetic algorithm involves three stages. Initially, in the crossover stage, two unrelated individuals are mated in which their offspring inherit parental features. After this, some offspring get mutated, and the genes may change randomly. Finally, the selection of the offspring of the present generation occurs based on the individual’s fitness: thus, solutions better suited to their environment reproduce, whereas poorer-suited ones die.
One of the implementations of GA is DOCK, whose purpose is to dock the active site of the entire ligand or only a rigid fragment. Genetic Optimisation for Ligand Docking (GOLD) is an automated docking based on how ligands bind to proteins by allowing complete acyclic ligand flexibility, partial cyclic ligand flexibility and partial protein flexibility. The possibility of a generated complex is evaluated by maximising the ligand-target hydrogen bonds 34. GOLD has mostly shown better accuracy than other methods.
Another genetic algorithm-based flexible docking program is the AutoDock program, which allows favourable phenotypic features to become inheritable. The AutoDock routine firstly plots an interaction energy map by using several probe atoms, and a subsequent search evaluates the binding energies using the maps. The latest version of AutoDock is AutoDock4.2 which is a two-point attractor approach. The approach penalises newly formed structures in which the sources are mispositioned. These tools support small binding sites and opened cavities and predict well even when the ligand is hydrophobic. This program only explores the torsional degrees of freedom, holding bond angles and constant bond lengths 35. The uncertainty of convergence in the outcomes is the main limitation of this algorithm. It also fails to operate accurately when the ligand is highly flexible and polar.
GOLD and AutoDock tools almost share the same advantages and disadvantages. Unlike the former, the latter does not appropriately rank ligands in large cavities.
Fragment bond simulations between ligand-protein pairs
The fragment-based approach is used to detect the interaction spots in a ligand-protein pair by simulating the bonding between their fragments. It avoids the degree of freedom entirely. The process for this approach involves five main steps. First, a diverse and good degree of suitable fragment library must be organised. The second stage is the virtual screening of the established library. Due to the small size of the fragments, it is not possible to make accurate energy estimates. However, analysis of potential atomic interactions can aid in finding appropriate segments. Then, the selected segments are combined to form novel compounds. Strategies like fragment growing, fragment linking, and lead fragments help in this design phase. Next, the generated complexes are evaluated with their biological assays. The false results are computationally eliminated before this step. The last step confirms the binding and understands the mechanism 36.
Time dependent docking computer simulations
Molecular Dynamics Simulation is a time-dependent docking mechanism that predicts the conformational positions of a protein at a particular time. It uses Newton’s laws of motion and thermodynamics to understand the atomic changes that occur in a protein’s structure over time and evaluate parameters such as energy 37. This method allows structural flexibility and entropic effects. The forces between bonded and non-bonded atoms contribute. Non-bonded forces between compounds arise due to van der Waals interactions 38-40. Software tools like AMBER, Abolane, GROMACS, MOE and YASARA
The recognition from bound to unbound state is difficult at the molecular level in molecular dynamics. To overcome this limitation, a modified version of Molecular Dynamics, Supervised Molecular dynamics, is proposed. This methodology investigates ligand-receptor binding events irrespective of its starting position, the ligand’s chemical structure, and its receptor binding affinity. This process is done by considering unbiased MD simulations and exploring the different pathways of protein-peptide binding 41.
This time-dependent model of Molecular Docking, Molecular dynamics, proves advantageous as it forms a more stable docked complex. Despite providing more accurate conformation, they prove difficult over high-barrier energy conformation. Moreover, the computational cost and time involved are huge.
Table 2: Structure based drug design – Molecular Docking Software Techniques
| ALGORITHM USED | TOOLS/ PROGRAMS | DESCRIPTION |
| Monte Carlo Docking Algorithm | MCDOCK | Program for predicting the binding mode of a ligand automatically by allowing full flexibility of ligands |
| Monte Carlo Docking Algorithm | AutoDock | Molecular docking simulation software which is effective for protein-ligand docking. AutoDock Vina is a successor of this software. |
| Monte Carlo Docking Algorithm | RiboDock | Software that is used for docking small molecules against proteins and nucleic acids. Designed for high-throughput virtual screening. |
| Evolutionary/Genetic Docking Algorithm | Catalyst | Automated Docking tool that shows how ligands bind to proteins by allowing various kinds of flexibilities |
| Evolutionary/ Genetic Docking Algorithm | GLIDE | Docking program that predicts protein-ligand binding modes and ranks ligands by high-throughput virtual screening 42 |
| Incremental
construction Incremental Construction (Docking) |
FlexX | Automatic docking tool for flexible ligand |
| Evolutionary Programming
(Docking) |
EADock | Uses a hybrid evolutionary algorithm with two fitness functions 43. |
| Molecular Dynamic Simulation | GROMACS | Uses a hybrid evolutionary algorithm with two fitness functions 43. |
| Molecular Dynamic Simulation | PLUMED | Simulates proteins, lipids, and nucleic acids. Compatible with both CPUs and GPUs. |
The structure-based tools are described in Table 2. Despite proving advantageous by simulating 3D complexes formed through drug-target interaction, the interactions are not possible without the 3D structure of the target proteins. The docking mechanism is also redundant when the target information is not available.
Advent of Chemogenic Approaches
The evolution of artificial intelligence over the years has resulted in its wide application in the healthcare field. These approaches use the information on both protein and drug molecules and combine their spaces for accurate predictions 25. They can be further classified into 2 types: feature-based and similarity-based 44-48.
Classification models of feature-based approach
The first approach is feature-based classification models. The models are trained with drug target pairs encoded as feature vectors 49,50. The distinct features are obtained from the pool of molecular drug characteristics using methods such as Principal Component Analysis or correlation matrix, then passed to the machine learning models. The feature models are then used to predict chemical bonding in a new pair of drugs and targets 25.
These models have high computational costs due to the complex process involved. However, the prediction models require target information to be known 51. This approach can be classified into three main divisions: CNN or SVM Based methods, Ensemble methods and Miscellaneous techniques.
Forecasting complex formation using Convoluted Deep Neural networks
A convolutional neural network is a supervised model that consists of convolution, max pooling, fully connected and output layer. Feature extraction is a convolution layer with a rectified linear unit (ReLU) activation function. A max-pooling layer is added to reduce the dimension of features. Finally, the fully-connection and output sigmoid layers are used to classify the tasks.
One of the latest adaptations of CNN, DTI-CNN, is a heterogenous model that has a feature extractor, DAE-based feature selector and interaction predictor. Initially, the features of the drugs-protein complexes are extracted using Jaccard similarity coefficient and Random walk parameters with a restart algorithm. Secondly, the selective low-dimensional representation of drug and protein features using the denoising autoencoder (DAE) model. Finally, a CNN model predicts the interaction between a given pair of drugs and proteins 52,53.
Another latest CNN model, FRnet-DTI, is a deep learning tool that includes an auto-encoder used in feature manipulation and a CNN used as a classifier for drug target interaction prediction. The autoencoder model, which also uses convolution, extracts about 4096 features from the given set of features and feeds its output to the FRnet Predict model 54-57. SDnDTI is another modified CNN method that denoises DTI data and does not require additional high-SNR data for training. The multi-dimensional input data is divided into many subsets of six DWI volumes and transformed the subset to match the same diffusion encoding directions. Each DWI is denoised using a deep 3-dimensional CNN model. This denoising ability aids this model in preserving image sharpness and textural details in the results.
Despite providing correct predictions, the Convolutional neural network models must often be combined with other models or require an increase in layer depth for better precision. This behaviour increases their overall computational cost. When the raw features are trained by passing through layers in CNN, each layer retains only a per cent of the feature values. Since the raw features are coupled with unnecessary noises, the filtering at each layer may drop essential values.
Role of Support Vector Machines in DTI
Support Vector Machines (SVM) is a machine learning method where a linear separator segregates two different sets of points in multi-dimensional space to predict contrasting classes. One advantage of the model is that it can process multi-dimensional patterns and define the relationships among them 58. This method offers great precision in all computations and works efficiently. The model can be used for both classification and regression purposes. At the beginning of SVM models, the classification models formulated several drug-related features like molecular similarity and chemical structure into a matrix. This matrix was inputted as a kernel to predict the classes 59. SVM is mainly used in nonlinear QSAR and virtual screening. Along with predicting target-specific activity, this method can also predict multi-target mechanisms. Classifier SVM is a type of multi-target SVM which generates a sequence of final classifiers by storing the distinct features from previous predictions. SVM also predicts activity cliffs and MMP cliffs by a graph of reaction representations in drug interactions. This method identifies proteins active against such “orphan” targets using reference molecules from similar targets. It employs a similarity searching mechanism where ligands that are similar show similar behaviour 60. One of the major drawbacks of SVM is that the number of false positives returned is higher than in other methods.
Procuring unique drug features and predicting using Ensemble based methods.
Ensemble-based methods obtain the features of drugs and targets and train an ensemble of unrelated decision trees. While predicting the possibility of interaction between a drug target pair, they receive the resulting outputs of all the ensembles and statistically combine them to get the final decision 61. Each decision tree adopts a top-down approach where the head node is a root node. Random forest is an ensemble-based algorithm suitable for large datasets with multiple dimensions 62. It procures relative features and processes them for classification. This model mitigates the condition of overfitting. Overfitting happens when a model also learns the noise from the training data, which affects the model’s overall performance. This can result in the negative evaluation of newly encountered data 63-66.
However, ensemble-based techniques like Random Forest show better predictions than CNN. The feature ranking embedded in each ensemble helps in better classification and, thus, precise prediction. Nevertheless, these models fail to handle negative sampling, a condition that arises when there is no information on drug-ligand interaction. Further, models, such as decision trees and SVMs, have a significant bias for finding the dominant class and thus result in poor performance 67.
Similarity based methods
Current databases with drug-target details contain a small number of experimentally validated sets of the pairs. Thus, many drug target pairs for which the interactions are not present. Since feature-based methods can’t predict when there is no complete knowledge about receptors, similarity-based models were introduced. This prediction assumes that if two molecules have similar properties, they are likely to be linked. The models can be used especially when uncertain information on any molecules 68,69,70. Furthermore, these models give high prediction accuracies as they have developed kernels 25. Here, the similitude between two drugs is generally represented in matrices. Similarly, the relation between two targets can also be shown in matrix format.
Predicting molecule formation using neighbour interaction profiles
Neighbourhood methods specialise in predicting the interaction profiles of drugs and targets based on their nearest neighbours’ interaction information. Neighbourhood methods such as k nearest neighbour, k means clustering and k medoids clustering are algorithms that cluster together drug target pairs that show similar characteristics. Thus, these methods can extrapolate interactions for an unknown pair from the attributes of a known pair.
The weighted nearest neighbour algorithm, called WNN, is the nearest neighbourhood model that constructs an interaction score profile for a new drug compound using the information about known compounds in the dataset. To compute the possibility of new interaction, the chemical similarity of the new drug is compared with other known drug compounds and their corresponding profiles are considered to generate a score interaction profile for that drug compound. These methods are often used in combination with other classification/ prediction models for better performance.
Matrix Factorization mechanism
The matrix factorisation method captures the latent feature metrics for drugs and targets from the Drug Target Interaction matrix. It then combines the metrics to reconstruct the interaction matrix for prediction.
The initial approach towards matrix factorisation in drug interaction mechanisms was with the development of the Bayesian-based matrix formulation method. Kernelized Bayesian Matrix Formulation combines dimensionality reduction, matrix factorisation and binary classification methods. They factor in precompiled similarity in chemical structure and protein patterns to predict drug-target interaction networks where similar compounds interact with similar proteins 71-74. The projection of drugs and target proteins into a multi-dimensional space helps estimating the similarities and formulating the joint Bayesian probabilities to solve this binary class prediction problem 75. The MF method is integrated with the Nearest neighbourhood method to improve the prediction results further.
A heterogeneous matrix factorisation method, which combines weighted K nearest known neighbours (WKNKN) with graph regularised matrix factorisation (GRMF), is developed. To mitigate the issue of unknown interactions, a KNKN model is designed to add edges with intermediate interaction likelihood scores. The model gets the close neighbours of the target ligands to predict the interaction. The latter is a matrix factorisation technique with a graph form of regularisation which predicts and avoids overfitting simultaneously 76.
Drug – target side integrated prediction using local network models
Bipartite local models (BLMs) comprise of two kinds of prediction: drug side and target side prediction, and then join these predictions to get the final prediction scores for the drug target pair.
Despite having good performance, the classic BLM, can’t learn without training data and thus predict new compounds. Therefore, a procedure called neighbour-based interaction-profile inferring (NII) is integrated with this model to classify new interactions. The interaction information procured from this procedure is treated as label information and utilised for new candidates’ behaviour prediction 77-79.
The traditional BLM-based methods cannot correctly predict new drugs or targets without any known interactions available. This resulted in extending the existing model by adding preprocessing to prove effective in new candidate problems.
The Bipartite Local Model is combined with the self-training Support Vector Machine to achieve better prediction accuracy to discover interactions. Before the classification, to categorise interactions as negative or positive, a k-medoids clustering method is used. K-medoids is an unsupervised partitioning technique of clustering where the data set of n objects is split into k clusters. The number k of clusters equals the number of positive interactions. First, BLM for a drug is trained with the help of an interaction profile of the drug and a similarity matrix of target proteins. Next, the self-training SVM is used as a classifier that differentiates known interactions (positive) from unknown interactions (negative) where the target similarity acts as a kernel. Finally, the model predicts the probability that interaction can exist between a drug-target pair by considering the similarities between a target and a trained target 80. The major limitation is that BLM also suffers from bias.
Mapping protein and drug molecule connections through Network systems
Network-based models represent the topological structure of the chemicals and proteins where the connection between them is represented as edges. Depending on their position, their features can be further derived locally or globally.
A graph-based method, DTINet, predicts novel drug–target interactions by constructing a heterogeneous network which combines diverse drug-related information. This model aims to learn a low dimensional informative feature vector representation and project them from drug to target vector space scheme for them to make accurate predictions. The feature vectors visualise the topological properties of individual nodes in the heterogeneous network. This model is robust to noisy and high dimensional data as it is designed to learn low dimensional features. The geometric proximity of the feature vectors 81-85.
To consider multiple data sources, a network framework, EFMSDTI is proposed to predict the interactions. First, multi-source data of drugs and targets are combined or split by classifying multi-source data. When there are multiple kinds of networks in a category, whether to join them into one network is determined according to their contribution to DTI prediction. It includes topological graphs containing Drug-drug, Drug-disease, Drug-side Effect, Target-target, and Target-disease interactions and a semantic graph with Drug similarities and Target similarities. Then, the networks are combined to get the low-dimensional representations of compounds based on the Graphical neural network models. Finally, LightGBM, a tree-based algorithm that grows vertically, is used. It is a gradient-boosting framework. Its ability to handle multiple data and reduced computational cost make it a good preference for prediction 86.
These models are primarily precise as they hold the merit of encoding the internal chemistry of the biomolecule. These properties determine the structural aspects, such as dynamics and structural altercations. The graph-based models integrated with multi-task learning can also be applied for highly imbalanced sub-datasets and perform better than descriptor-based models. Building generalisable and robust graph models require large-scale, high-quality datasets. However, the datasets in the practical drug databases suffer from narrow chemical diversity and insignificant sample sizes. Moreover, the prediction performances for such models are not so convincing.
Network based deep learning methods to integrate proteins and drugs interactions
Network diffusion methods investigate graph-based techniques to influence propagation in drug–target networks and predict novel DTIs. These methods are efficient in finding and establishing unknown drugs to target interactions. As a result, they are often combined with other prediction models to achieve better accuracy 87-90. The integration of protein-to-protein interaction and drug-to-drug interaction has helped further improve the network models’ overall performance.
By giving the topological information of protein interaction interactome (PPI) and drug-to-drug interaction (DDI), a classic random forest algorithm predicts the DTI. With the help of these two interactions, the features of the drugs and targets are measured through weight function. However, this method has a drawback. It takes only the immediate neighbours into account. To mitigate this shortcoming, the Random walk restart algorithm is applied to both PPI and DDI networks separately. Then, with the final affinity scores from the RWR algorithm, the feature vectors are reweighted to generate negative drug-target interactions.
Conclusion
The development of effective computational models and accurate drug-target interaction prediction make a significant revolution in drug discovery research. The application of these innovative drug-target strategies overcomes the challenges in developing the lead therapeutic candidates. In this review, we have adopted a hierarchical classification scheme comprising three main categories: Ligand-based models, structure-based models, and machine learning models. Ligand-based methodologies focus on forecasting drug-target binding by analysing structural similarities between them. CADD models, like virtual simulations, can help identify the interaction in drug-target pairs when the conformational properties are present. Lastly, the machine learning-based method also enhances the prediction and favours accurate results. Different algorithms need to be employed and integrated to achieve greater precision and effective forecasts based on the specificity of the algorithms. Moreover, the multi-target multi-drug models could be a promising tool for drug repurposing against most life-threatening diseases. So, the invention and application of these technically advanced drug-target predictions help a successful procession of drug development stages and serve mankind.
Machine learning has undoubtedly improved prediction outcomes in DTI prediction, but there remains scope for further development. The richness of data available in drug databases presents both opportunities and challenges. Data cleaning and sampling are labour-intensive tasks that necessitate much more advanced tools. The limited availability of some drug target pairs in databases can result in the underperformance of supervised machine-learning models. Thus, more semi-supervised models must be equipped to provide more comprehensive insights into the interaction despite the scarcity of labelled data. Thus, the further advancement of machine learning technology holds promise in advancing our understanding of complex biological systems and facilitating drug discovery processes efficiently.
Acknowledgement
Bharath Institute of Higher Education and Research (BIHER), Chennai, India. University of Massachusetts, Amherst, Massachusetts, USA.
Conflict of Interests
The authors declare that they have no conflicting interests.
Funding Source
There is no funding source for this research.
References
- Ali Ezzat, Min Wu, Xiao-Li Li, Chee-Keong Kwoh. Computational prediction of drug–target interactions using chemogenomic approaches: an empirical survey. Brief Bioinform., 2019;20(4):1337-1357.
- Shi J-Y, Yiu S-M. SRP: a concise non-parametric similarity-rank-based model for predicting drug-target interactions. In: IEEE International Conference on Bioinformatics and Biomedicine (BIBM). Washington, DC, USA: IEEE., 2015; 1636–41.
- Zong N, Kim H, Ngo V, et al. Deep mining heterogeneous networks of biomedical linked data to predict novel drug-target associations. Bioinformatics., 2017; b33:2337–44.
- Batool M, Ahmad B, Choi S. A Structure-Based Drug Discovery Paradigm. Int J Mol Sci., 2019;20(11):2783.
- Olivecrona, M., Blaschke, T., Engkvist, O. Chen, H. Molecular de-novo design through deep reinforcement learning. J. Cheminform., 2017; 9(1):48.
- Pereira, J.C., Caffarena, E.R., dos Santos, C.N. Boosting docking-based virtual screening with deep learning. J. Chem. Inf. Model., 2016; 56:2495–2506.
- Bhagat Rani, Butle Santosh, Khobragade Deepak, Wankhede Sagar, Prasad Chandani, Mahure Divyani, Armarkar Ashwini. Molecular Docking in Drug Discovery. J Pharm Res Int., 2021;33:46-58.
- Bian Yuemin, Xie Xiangqun. Computational Fragment-Based Drug Design: Current Trends, Strategies, and Applications. AAPSJ., 2018;20(3):59.
- Chen R, Liu X, Jin S, Lin J, Liu J. Machine Learning for Drug-Target Interaction Prediction. Molecules., 2018;23(9):2208.
- Hartenfeller, M. & Schneider, G. De novo drug design. Methods in molecular biology., (2011); 672: 299–323.
- Richardson, Janes S & Richardson, David C. The de novo design of protein structures. Trends in Biochemical Sciences., 1989; 14(7): 304-309.
- Collin C.B, Gebhardt T, Golebiewski M, Karaderi T, Hillemanns M, Khan F.M, Salehzadeh-Yazdi A. Kirschner M, Krobitsch S. EU-STANDS4PM consortium: Kuepfer L. Computational Models for Clinical Applications in Personalized Medicine—Guidelines and Recommendations for Data Integration, J Pers Med., 2022;12:166.
- Dar A, Ayaz. Molecular Docking: Approaches, Types, Applications and Basic Challenges. J Anal Bioanal Tech., 2017;8:356.
- Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des.,2010 ;24(5):417-22.
- Deore A, Dhumane J, Wagh R, Sonawane R. The Stages of Drug Discovery and Development Process. Asian J Pharm Res Dev., 2019;7(6):62-67.
- Target discovery. Nature Reviews Drug Discovery.,2003; 2: 831–838.
- TerstappenG,Schlüpen,C,RaggiaschiR,GaviraghiG.Target deconvolution strategies in drug discovery. Nature Reviews Drug Discovery., 2007; 6(11):891-903.
- Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nature Reviews Drug Discovery, 2006; 5:821-834
- Dias Raquel, De Azevedo, Jr Walter. Molecular Docking Algorithms. Curr Drug Targets., 2009;9:1040-1047.
- Wang, Renxiao & Liu, Liang & Lai, Luhua & Tang, Youqi. SCORE: A New Empirical Method for Estimating the Binding Affinity of a Protein-Ligand Complex. Journal of Molecular Modeling.,1998;4: 379-394.
- Liu, Ming & Wang, Shaomeng. MCDOCK: A Monte Carlo simulation approach to the molecular docking problem. Journal of Computer-AidedMolecularDesign.,1999:13:435-451.
- Durrant J.D, McCammon J.A. Molecular dynamics simulations and drug discovery. BMC Biol., 2011;9:71.
- Ewing TJ, Kuntz I D. Critical evaluation of search algorithms for automated molecular docking and database screening. J Comput Chem., 1997;18:1175-1189.
- Ezzat Ali, Zhao Peilin, Wu Min li, Xiaoli Kwoh, Chee-Keong. Drug-Target Interaction Prediction with Graph Regularized Matrix Factorization. IEEE/ACM Trans Comput Biol Bioinform., 2016;14(3):646-656.
- Ezzat A, Wu M, Li XL, Kwoh CK. Computational prediction of drug-target interactions using chemogenomic approaches: an empirical survey. Brief Bioinform., 2019;20(4):1337-1357.
- Farshid Rayhan, Sajid Ahmed, Zaynab Mousavian, Dewan Md Farid, Swakkhar Shatabda. FRnet-DTI: Deep convolutional neural network for drug-target interaction prediction. Heliyon., 2020;6(3): e03444.
- Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules., 2015;20(7):13384-13421.
- Firoozbakht Forough, Rezaeian Iman, Rueda Luis, Ngom Alioune. Computationally repurposing drugs for breast cancer subtypes using a network-based approach. BMC Bioinformatics., 2022;23:143.
- Gareth Jones, Peter Willett, Robert C Glen, Andrew R Leach, Robin Taylor. Development and validation of a genetic algorithm for flexible docking. J Mol Biol., 1997;267(3):727-748.
- Grosdidier A, Zoete V, Michielin O. EADock: docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins., 2007;67(4):1010-1025.
- G¨onen, Mehmet. Predicting drug-target interactions from chemical and genomic kernels using Bayesian matrix factorization. Bioinformatics., 2012;28:2304-2310.
- Gelfand A.E., Smith A.F.M.. Sampling-based approaches to calculating marginal densities, J. Amer. Statist. Assoc., 1990;85: 398-409.
- Neal R.M.. , Bayesian Learning for Neural Networks, 1996New York, NYSpringer
- Hao Ding, Ichigaku Takigawa, Hiroshi Mamitsuka, Shanfeng Zhu. Similarity-based machine learning methods for predicting drug–target interactions: a brief review. Brief Bioinform., 2014;15(5):734–747.
- Janardhanan Padmavathi, Heena L, Sabika Fathima. Effectiveness of Support Vector Machines in Medical Data mining. J Commun Softw Syst., 2015;11:25-30
- Jian-Ping Mei, Chee-Keong Kwoh, Peng Yang, Xiao-Li Li, Jie Zheng, Drug–target interaction prediction by learning from local information and neighbors. Bioinformatics, 2013;29(2):238–245.
- Keum Jongsoo, Nam Hojung, SELF-BLM: Prediction of drug-target interactions via self-training SVM.PLoS ONE., 2017;12(2): e0171839.
- Layla Abdel-Ilah, Elma Veljovic, Lejla Gurbeta, Almir Badnjevic, Applications of QSAR Study in Drug Design. International Journal of Engineering Research and Technology, 2017; 6(6):582-587.
- Veerasamy R, Rajak H, Jain A, Sivadasan S, Varghese CP, Agrawal RK.Validation of QSAR Models – Strategies and Importance. InternationalJournal of Drug Design and Discovery. 2011; 2(3), 511-519.
- Van de Waterbeemd H, Testa B, Folkers G. Computer-Assisted LeadFinding and Optimization. VHChA and VCH, Basel, Weinheim. 1997;9-28.
- Lin, Xiaoqian Li, Xiu Lin, Xubo, A Review on Applications of Computational Methods in Drug Screening and Design. Molecules, 2020;25:1375.
- Liu M, Wang S, MCDOCK: A Monte Carlo simulation approach to the molecular docking prob. J Comput Aided Mol Des., 1999;13: 435–451.
- Luo Y, Zhao X, Zhou J, et al, A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat Commun., 2017;8: 573.
- Lu Y, Guo Y, Korhonen A, Link prediction in drug-target interactions network using similarity indices. BMC Bioinformatics, 2017;18:39.
- Yamanishi Y, Araki M, Gutteridge A, Honda W, Kanehisa M. Prediction ofdrug–target interaction networks from the integration of chemical andgenomic spaces. Bioinformatics. 2008;24(13):232–40.
- Nakhjavani M, Hamidi S, Esteghamati A, Abbasi M, Nosratian-Jahromi S,Pasalar P. Short term effects of spironolactone on blood lipid profile: a3-month study on a cohort of young women with hirsutism. Br J ClinPharmacol. 2009;68(4):634–7.
- Ding H, Takigawa I, Mamitsuka H, Zhu S. Similarity-based machinelearning methods for predicting drug–target interactions: a brief review.Brief Bioinform. 2014;15(5):734–47.
- Chen X, Yan CC, Zhang X, Zhang X, Dai F, Yin J, Zhang Y. Drug–targetinteraction prediction: databases, web servers and computationalmodels. Brief Bioinform. 2015;17(4):696–7.
- Makhoba X H, Jr C V, Mosa R A, D Viegas F P, Pooe, O J, Potential Impact of the Multi-Target Drug Approach in the Treatment of Some Complex Diseases. Drug Design, Development and Therapy, 2020; 14: 3235-3249.
- Medina-Franco JL, Giulianotti MA, Welmaker GS, Houghten RA. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov Today. 2013;18(9–10):495–501.
- Ramsay RR, Popovic-Nikolic MR, Nikolic K, Uliassi E, Bolognesi ML. A perspective on multi-target drug discovery and design for complex diseases. Clin Transl Med. 2018;7(1):3.
- Napolitano F, Zhao Y, Moreira VM, et al, Drug repositioning:a machine-learning approach through data integration. J Cheminform, 2013;5:30.
- Mukhopadhyay, Tapas & Sasaki, Jiichiro & Ramesh, Rajagopal & Roth, Jack. Mebendazole elicits a potent antitumor effect on human cancer cell lines both. Clinical cancer research : an official journal of the American Association for Cancer Research., 2002; 8: 2963-9.
- Patel L, Shukla T, Huang X, Ussery DW, Wang S, Machine Learning Methods in Drug Discovery. Molecules, 2020; 25(22):5277.
- Reker, D.; Bernardes, G.J.L.; Rodrigues, T. Computational advances in combating colloidal aggregation in drug discovery. Nat. Chem. 2019; 11: 402–418.
- Mohanty, S.; Rashid, M.H.A.; Mridul, M.; Mohanty, C.; Swayamsiddha, S. Application of Artificial Intelligencein COVID-19 drug repurposing. Diabetes Metab. Syndr. 2020; 14: 1027–1031.
- Kowalewski, J.; Ray, A. Predicting novel drugs for SARS-CoV-2 using machine learning from million chemical space. Heliyon 2020;6:e04639.
- Paul D, Sanap G, Shenoy S, Kalyane D, Kalia K, Tekade R K, Artificial intelligence in drug discovery and development. Drug Discovery Today, 2021;26(1): 80-93.
- Peng Jiajie, Li Jingyi, Shang Xuequn, A learning-based method for drug-target interaction prediction based on feature representation learning and deep neural network. BMC bioinformatics, 2020;21(13):394.
- Pliakos, Konstantinos, Vens, Celine, Drug-target interaction prediction with treeensemble learning and output space reconstruction. BMC Bioinformatics., 2020; 21:40.
- Prada-Gracia D, Huerta-Y´epez S, Moreno-Vargas LM, Application of computational methods for anticancer drug discovery, design, and optimization. Bol Med Hosp Infant Mex., 2016;73(6):411-423.
- Qing X, Lee XY, De Raeymaecker J, Tame J, Zhang K, De Maeyer M, Voet A, Pharmacophore modeling: advances, limitations, and current utility in drug discovery. Journal of Receptor, Ligand and Channel Research, 2014;7:81-92.
- Rajan Chaudhari, Long Wolf Fong, Zhi Tan, Beibei Huang, Shuxing Zhang, An upto-date overview of computational polypharmacology in modern drug discovery. Expert Opinion on Drug Discovery. ,2020;15:1025-1044.
- Anighoro A, Bajorath J, Rastelli G. Polypharmacology: challenges and opportunities in drug discovery.J Med Chem.2014; 9;57(19):7874–87.
- Labrijn AF, Janmaat ML, Reichert JM, et al. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019; 18(8):585–608.
- Tan Z, Chaudhai R, Zhang S. Polypharmacology in Drug Development:A Minireview of Current Technologies. ChemMedChem., 2016; 20:11(12):1211–8.
- Repasky MP, Shelley M, Friesner RA, Flexible ligand docking with Glide. Curr Protoc Bioinformatics, Wiley, 2007. doi: 10.1002/0471250953.bi0812s18. PMID: 18428795.
- Rodr´ıguez-P´erez R, Bajorath J, Evolution of Support Vector Machine and Regression Modeling in Chemoinformatics and Drug Discovery. J Comput Aided Mol Des., 2022;36:355–362.
- Zernov VV, Balakin KV, Ivaschenko AA, Savchuk NP, Pletnev IV. Drug discovery using support vector machines. The case studies of drug-likeness, agrochemical-likeness, and enzyme inhibition predictions. J Chem Inf Comput Sci.,2003;43(6):2048-56.
- Ekins S, Reynolds RC, Kim H, Koo MS, Ekonomidis M, Talaue M, Paget SD, Woolhiser LK, Lenaerts AJ, Bunin BA, Connell N. Bayesian models leveraging bioactivity and cytotoxicity information for drug discovery. Chem Biol.2013;20:370–378.
- Sabbadin Davide, Salmaso Veronica, Sturlese Mattia, Moro Stefano, Supervised Molecular Dynamics (SuMD) Approaches in Drug Design.Methods Mol Biol., 2018;1824:287-298.
- Cuzzolin A, Sturlese M, Deganutti G et al Deciphering the complexity of ligand-protein recognition pathways using supervised molecular dynamics (SuMD) simulations. J Chem Inf Model., 2016; 56(4):687–705.
- Harvey MJ, Giupponi G, Fabritiis GD ACEMD: accelerating biomolecular dynamics in the microsecond time scale. J Chem Theory Comput.,2009;5(6):1632–1639.
- Petros AM, Nettesheim DG, Wang Y et al.Rationale for Bcl-xL/bad peptide complex formation from structure, mutagenesis, and biophysicalstudies.ProteinSci.,2000;9(12):2528–2534.
- Schlander M, Hernandez-Villafuerte K, Cheng CY, Mestre-Ferrandiz J, Baumann M, How Much Does It Cost to Research and Develop a New Drug? A Systematic Review and Assessment. Pharmacoeconomics, 2021;39(11):1243-1269.
- Singh Sakshi, Bani Baker Qanita, Singh Dev Bukhsh, Molecular docking and molecular dynamics simulation. Bioinformatics, Methods and Applications, Elsevier, 2021;291- 304.
- Wang C, Zhang J, Chen P, Wang B, Predicting Drug-Target Interactions Based on the Ensemble Models of Multiple Feature Pairs,J Mol Sci., 2021;22(12):6598.
- Li, Z.R, Lin, H.H, Han, L.Y et al, PROFEAT: A web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res., 2006;34:W32–37.
- Zhang, P,Tao, L. Zeng, X et al, PROFEAT Update: A Protein Features Web Server with Added Facility to Compute Network Descriptors for Studying Omics-Derived Networks. J. Mol. Biol., 2017;429: 416–425.
- Wang, Y. Deng, Y. Wu, B. Kim, D.N.Lebard, D. Wandschneider, M. Beachy, R.A. Friesner, R. Abel, Accurate modeling of scaffold hopping transformations in drug discovery. J Chem Theor Comput., 2017;13 : 42-54.
- Zaynab Mousavian, Ali Masoudi-Nejad, Drug–target interaction prediction via chemogenomic space: learning-based methods, Expert Opin Drug Metab Toxicol., 2014;10(9): 1273-1287.
- Masoudi-Nejad A, Mousavian Z, Bozorgmehr JH. Drug-target and disease networks: polypharmacology in the post-genomic era. In Silico Pharmacol., 2013;1(1):1-4.
- Butina D, Segall MD, Frankcombe K. Predicting ADME properties in silico: methods and models. Drug Discov Today., 2002;7(11):S83-8.
- Byvatov E, Fechner U, Sadowski J, et al. Comparison of support vector machine and artificial neural network systems for drug/nondrug classification. J Chem Inf Comput Sci., 2003;43(6):1882-9.
- Cheng AC, Coleman RG, Smyth KT, et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol., 2007;25(1):71-5.
- Zhang W, Bell E.W, Yin M, et al, EDock: blind protein–ligand docking by replicaexchange monte carlo simulation, J Cheminform., 2020;12:37.
- Zhang Yuan , Wu Mengjie , Wang Shudong , Chen Wei. EFMSDTI: Drug-target interaction prediction based on an efficient fusion of multi-source data. Frontiers in Pharmacology, 2022; 13: 1009996.
- Liu, T., Lin, Y., Wen, X., Jorissen, R. N., and Gilson, M. K. (2007). BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35:D198–201.
- Luo, Y., Zhao, X., Zhou, J., Yang, J., Zhang, Y., Kuang, W., et al. (2017). A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Commun.2017; 8:
573. - Olayan, R. S., Ashoor, H., and Bajic, V. B. (2018). Ddr: Efficient computational method to predict drug-target interactions using graph mining and machine learning approaches. Bioinformatics, 20
18; 34:1164–1173.

This work is licensed under a Creative Commons Attribution 4.0 International License.



