

How to Cite | Publication History | PlumX Article Matrix
Samiksha S. Amrutkar* , Shivam K. Navale, Deepak S. Bhambere, Pooja Birari , Shweta Shevatkar , Sapana P. Ahirrao , Sanjay J. Kshirsagar , Sameer Vijay Darade
, Shivam K. Navale, Deepak S. Bhambere, Pooja Birari , Shweta Shevatkar , Sapana P. Ahirrao , Sanjay J. Kshirsagar , Sameer Vijay Darade
Department of Pharmaceutics, METs Institute of Pharmacy, Nashik Maharashtra, India
Corresponding Author E-mail: amrutkarsamiksha@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3298

ABSTRACT: This study aimed to enhance the solubility and consequent bioavailability of Loratadine, a widely used medication for seasonal allergic rhinitis and chronic idiopathic urticaria. The approach involved complexation with β-cyclodextrin to improve solubility and the formulation of fast-disintegrating tablets via the sublimation method. Crosspovidone acted as a superdisintegrant, while camphor served as a sublimating agent. Methodology: Pre-compression parameters were assessed to ensure adequate flow properties, while post-compression parameters were evaluated against IP acceptable limits for variations in hardness, weight, wetting time, friability, drug release, and disintegration time, drug content, FTIR, and DSC. Dissolution studies were conducted in 0.1N HCL, and statistical analysis employed a central composite design for optimization. Results: Formulation B14 emerged as the optimized batch, demonstrating favourable characteristics including wetting time, in vitro drug release, drug content, and disintegration time. Analysis of stability conducted in accordance with ICH guidelines affirmed the formulations' stability. Conclusion: The developed fast-disintegrating tablet formulation of Loratadine exhibited enhanced solubility, potentially translating to improved bioavailability. These advancements suggest increase efficacy and improved patient compliance in the management of allergic rhinitis and urticaria.
KEYWORDS: Central Composite Design; Disintegration time; Dissolution; Fast disintegrating tablet; Loratadine
Download this article as:| Copy the following to cite this article: Amrutkar S, Navale S. K, Bhambere D. B, Birari, P, Shevatkar S, Ahirrao S. P, Kshirsagar S. J, Darade S. V. Development and Assessment of Loratadine Fast-Disintegrating Tablets via β-Cyclodextrin Complexation with Superdisintegrants. Biotech Res Asia 2024;21(3). |
| Copy the following to cite this URL: Amrutkar S, Navale S. K, Bhambere D. B, Birari, P, Shevatkar S, Ahirrao S. P, Kshirsagar S. J, Darade S. V. Development and Assessment of Loratadine Fast-Disintegrating Tablets via β-Cyclodextrin Complexation with Superdisintegrants. Biotech Res Asia 2024;21(3). Available from: https://bit.ly/4eI5jho |
Introduction
The compound, known as Loratadine, chemically corresponds to the 2-(8-chloro-5, Six-dihydro-11H-benzo [5, 6] cyclohepta [1, 2-b] pyridin-11-ylidene)-1-piperidine with carboxylic acid ethyl ester. It possesses pKa values around 5.25 and demonstrates minimal solubility in water (0.0004-0.006 mg/ml), while displaying solubility in 0.1N HCl.1
The Biopharmaceutical Classification System (BCS) offers a structured approach to categorize drugs based on their solubility, permeability, and dissolution properties concerning their dosage form. According to this system, Loratadine is classified as a Class II drug, indicating that its dissolution rate may hinder its absorption. Its limited solubility in water leads to inconsistent dissolution in both stomach and intestine fluids, affecting its absorption. Consequently, the absorption rate and extent are largely determined by its dissolution in the digestive tract. Improving the solubility and dissolution of poorly water-soluble drugs possess a significant obstacles in drug development. Multiple strategies have been proposed to tackle this problem, include solubilization, salt formation, micronization, polymer complexation, altering physical forms, solid dispersions, complexation, and utilizing hydrophilic carriers.2
Inclusion Complex with Cyclodextrin3
Cyclodextrin, cyclic oligomers of glucose, exhibit the capacity to combine with both small molecules and parts of greater compounds to generate water-soluble inclusion complexes. These biocompatible cyclic oligosaccharides are distinguished by their immunologically inert nature and low toxicity profiles in both animal and human subjects. Within pharmaceutical contexts, cyclodextrins serve multifaceted roles, notably in augmenting drug bioavailability. A noteworthy application involves the utilization of cyclodextrin-incorporating polymers, which confer distinct advantages in the delivery of nucleic acids.
Methods of preparation of cyclodextrin inclusion complex
Grinding
Extrusion
Kneading
Solid dispersion
Slurry complexation
Orally Disintegrating Tablets (ODTs) represent a novel drug delivery modality designed to rapidly disintegrate within the oral cavity upon ingestion, obviating the need for chewing and the use of water, which sets them apart from conventional immediate-release oral solid dosage forms and other delivery systems. This formulation offers notable benefits, particularly for acute conditions, facilitating convenient administration anytime and anywhere symptoms arise, thereby enhancing patient adherence. Additionally, for chronic conditions, ODTs are anticipated to bolster patient compliance. Key advantages of ODT drug delivery include enhanced ease of swallowing for patients and the convenience of medication ingestion without the necessity of water.4 Pediatric and geriatric populations, along with individuals who are unwell and confined to a supine position in bed, as well as those who are frequently on the move without access to water, encounter significant challenges in swallowing tablets. Innovative oral drug delivery systems that rapidly dissolve or disperse within seconds upon placement in the mouth, without the need for water, offer a promising solution to alleviate the difficulties associated with tablet swallowing.4 The ideal characteristics for Fast Disintegrating Tablets (FDTs) include a pleasant mouthfeel, minimal to without a mark beneath within the mouth with respect to delivery, and the ability to dissolve or disintegrate within seconds without the need for water to aid swallowing.5, 6
In the current research, efforts have been directed towards formulating fast-dissolving tablets of Loratadine with enhanced solubility achieved through complexation with Beta-cyclodextrin, utilizing the kneading method. The tablets were prepared using the sublimation method, chosen for its superior performance compared to direct compression methods, notably yielding faster disintegration times. This formulation strategy leveraged the action of superdisintegrants like crosspovidone and sublimating agents such as camphor. The study investigated the effects of sublimated tablets (comprising the drug, Beta-cyclodextrin, and sublimating agent) on various parameters including stability, drug content, wetting time and disintegration time, in-vitro release under specified conditions.
Materials and Methods
Materials:
A gift sample of the drug Loratadine was provided by Glenmark Pharmaceuticals Ltd., R&D – Taloja. Crosspovidone, β-Cyclodextrin, Camphor, Talc, Magnesium Stearate, and Sodium Saccharine were sourced from METs Institute of Pharmacy, ensuring pharmaceutical and analytical grade quality. The research utilized instruments including a Weighing Balance, UV Spectrophotometer (Shimadzu – 1800), and a dissolution test apparatus (ELECTROLAB) to conduct the experiments.
Methods
Characterization and identification of drug
Physical characterization of drug
The physical characteristics of the drug were evaluated by placing it on a clean surface and observing it with the naked eye. Observations were recorded accordingly.
Determination of melting point7
The stage of Melting Apparatus was employed to determine the melting point of Loratadine through the following steps
A small quantity of Loratadine crystals was carefully inserted into a one-end sealed thin-walled capillary tube.
The capillary tube, along with a thermometer, was positioned within the Melting Point Apparatus.
The sample was subjected to control heating to induce melting.
The temperature at which Loratadine began to melt was recorded.
Solubility8
10 mg of Loratadine, precisely measured, were dissolving in ten milliliters of filtered water and 10 ml of 0.1N HCL, separately, in a vial. After that, these solutions were allowed to remain for an entire day at room temperature on a magnetic stirrer. Whatman filter paper was used to filter the samples after the specified period of time, and the filtrates were analysed for absorbance.
Preparation of calibration curve: 7
Calibration curve in 0.1N HCL
A flask with a volume of 100 mL was filled with 10 mg of Loratadine after it had been precisely measured. After that, the volume was adjusted using 0.1 N HCL to provide a standard stock solution with a 100 μg/mL concentration. A working standard solution containing 10 μg/mL worked together via extraction 1 mL from the normal stock solution, which has been then diluted with 0.1 N HCL to a final concentration of 10 mL. Subsequent dilutions were performed to achieve a concentration range spanning from 10 μg/mL to 100 μg/mL in 0.1 N HCL as the solvent.
Calibration curve in methanol:
After precisely weighing 10 mg of Loratadine, the level of volume was increased to 100 mL with methanol to generate a standard stock solution with a concentration of 100 μg/mL. With a concentration and a working standard solution of 10 μg/mL was obtained by using 1 mL of this standard stock mixture and diluting it out with methanol to a final volume of 10 mL.
Further dilutions were then carried out to achieve a concentration range spanning from 10 μg/mL to 100 μg/mL, with methanol serving as the solvent.
IR studies9
The pure drug’s infrared spectrum, 1:1 inclusion complex, and powder formulation were obtained using an Infrared spectrophotometer with KBr pellets as the medium for recording.
DSC studies9
DSC (Differential Scanning Calorimetry) studies were conducted on both pure Loratadine and the optimized formulation batch, such as F14. Approximately 5 mg of accurately weighed Loratadine and the optimized formulation were subjected to DSC analysis using an automatic thermal analyser system.
Preparation of Inclusion complex of Loratadine with β-Cyclodextrin10
Kneading method
Loratadine (100 mg) and β-Cyclodextrin (equivalent to 100 milligram of drug) were combined and placed into a mortar. Gradually, 2-3 the mixture was mixed with a few drops of distilled water, while vigorously mixing to achieve a uniform paste consistency. For 10 to 15 minutes, the paste was vigorously mixed to continue keeping things constant throughout the entire procedure. The mixture was then dried in a thermal chamber at 40°C. The bulk was crushed to obtain a powdered form.
Table 1: Composition of inclusion complexes
| Sr no. | Drug : Polymer | Ratio |
| 1. | Loratadine : β-Cyclodextrin | 1:1 |
| 2. | Loratadine : β-Cyclodextrin | 1:2 |
Evaluation of inclusion complexes:
Analysis of drug content in inclusion complexes11
Method – Utilizing a double beam UV-visible spectrophotometer made by Shimadzu Co. in Japan, the amount of Loratadine in each inclusion complex was ascertained. 0.1N HCL and 100 milliliters of distilled water were used to dissolve the powder and equated to 10 milligrams of Loratadine was used to calculate the percent medication content of each inclusion complex. After that, the mixture was passed through Whatman filter paper number 42. The absorbance was measured at 273 nm and 275 nm while the required dilutions were being prepared.
Saturation solubility8
Loratadine solubility both in its inclusion complexes and as a bulk drug was assessed in both distilled water and 0.1N HCL.
Method – After the preparation of inclusion complexes equal to 10 mg of Loratadine, 10 mL of the equivalent medium was put into every vial. After that, the vials were left at room temperature for a whole day on a magnetic stirrer. Following this incubation time, samples were filtered using Whatman filter paper No. 42, and measurements of absorbance were taken.
Dissolution studies9
The drug’s dissolving investigations on inclusion complexes and pure drug (equal to 10 milligrams of drug) loaded in empty capsule were performed in distilled water and 0.1N HCL by using Dissolution Test Apparatus TDT-08 Electrolab, Mumbai.
Dissolution medium – Distilled water and 0.1N HCL (900ml)
Speed – 75 rpm
Temperature – 370C ± 0.50C
Apparatus – USP II (Rotating paddle type)
Method
During the process of dissolution analysis, a 10 mL A sample was retrieved out at intervals of 5, 10, 15, 20, 25, and 30. It was then shifted out for a comparable quantity of entirely fresh medium as the material that was removed was filtered using Whatman filter paper number 42. At 275 nm, spectrophotometric analysis was performed on the filtered samples.
Formulation Design of Fast Disintegrating Tablets by Direct Compression Method: Incorporation of Sublimating Agent and Superdisintegrant
The tablet formulation comprised Loratadine as the active ingredient, β-Cyclodextrin as a polymer, crospovidone as a superdisintegrant, and camphor as a sublimating agent. Additional excipients included lactose as a diluent, sodium saccharine, talc, and magnesium stearate. The tablet weight remained consistent across batches, each containing 30 tablets, prepared via direct compression using a single punch hand-operated tablet compression machine. The compressed tablets underwent sublimation in an oven at 50°C for 15 minutes.
Table 2: Investigating range of variables
| Sr no. | Factors | Low level (%) | High level (%) |
| 1. | Superdisintegrant | 5% | 10% |
| 2. | Sublimating agent | 10% | 20% |
Table 3: Design of formulation of preformulation batches of fast disintegrating tablet by Central Composite Design
| No. | Composition | Quantity (mg) | ||||||||
| B1 | B2 | B3 | B4 | B5 | B6 | B7 | B8 | B9 | ||
| 1. | Loratadine Complexation | 69.28 | 69.28 | 69.28 | 69.28 | 69.28 | 69.28 | 69.28 | 69.28 | 69.28 |
| 2. | Crospovidone | 7.5 | 15 | 7.5 | 15 | 5.94 | 16.56 | 11.25 | 11.25 | 11.25 |
| 3. | Camphor | 15 | 15 | 22.5 | 22.5 | 18.75 | 18.75 | 13.44 | 24.06 | 18.75 |
| 4. | Lactose | 52.22 | 44.72 | 44.72 | 37.22 | 50.03 | 39.41 | 50.03 | 39.41 | 44.72 |
| 5. | Sodium Saccharine | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 |
| 6. | Talc | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| 7. | Magnesium Stearate | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 |
| Over all | 150 | 150 | 150 | 150 | 150 | 150 | 150 | 150 | 150 | |
| No. | Ingredients | Quantity | |||
| B10 | B11 | B12 | B13 | ||
| 1. | Loratadine Complexation | 69.28 | 69.28 | 69.28 | 69.28 |
| 2. | Crospovidone | 11.25 | 11.25 | 11.25 | 11.25 |
| 3. | Camphor | 18.75 | 18.75 | 18.75 | 18.75 |
| 4. | Lactose | 44.72 | 44.72 | 44.72 | 44.72 |
| 5. | Sodium Saccharine | 1.5 | 1.5 | 1.5 | 1.5 |
| 6. | Talc | 3 | 3 | 3 | 3 |
| 7. | Magnesium Stearate | 1.5 | 1.5 | 1.5 | 1.5 |
| Over all | 150 | 150 | 150 | 150 | |
Using the direct compression approach, fast-disintegrating Loratadine tablets were developed by combining the sublimating agent camphor and the superdisintegrant crospovidone. To get the optimal potential friability, thickness, hardness, disintegration time, and drug release, several formulations were developed. The formulation of the tablets involved a thorough mixing of Loratadine, corresponding to 10 mg β-Cyclodextrin complexation, with a pestle and glass mortar. Sublimating agent, diluent, and superdisintegrant were then added to the powder mixture according to with the formulae shown in Table 5. Subsequently, talc, sodium saccharine, and magnesium stearate used as lubricant. After passing the mixture through sieve number 80, the crushed tablets were sublimated for 15 minutes at 50°C in an oven.
Evaluation of pre-compression parameters of drug and excipients12
Angle of repose
The angle of repose is the maximum angle that can occur between a powder pile’s surface and a horizontal plane. The funnel method was used to calculate the angle of repose. The angle of repose was determined using the following formula:
Tan θ= h/r
Bulk density
The following formula was followed to get the bulk density:
Weight of the powder (M) / Volume of the packing equates to bulk density (ρ0).
Tapped density
The following formula was used to calculate the tapped density:
Weight of the powder (M) / Volume of the tapped packing equates to Tapped Density (ρ0).
Compressibility Index
Calculate percent compressibility using the following formula
Index of Compressibility (%) = [(Tapped Density-Bulk Density) * 100]/ Tapped Density
Hausner’s Ratio
Hausner’s ratio= (TD) / (BD)
Physical Parameters of Fast Disintegrating Tablet: 12
Thickness
It’s expressed in millimeters. An Advance Dial Caliper was used to measure the tablet thickness.
Hardness
The hardness of a tablet determines how resistant it is to breaking during shipping, storage, transportation, and handling prior to use. The Monsanto hardness tester was used to determine the 6 number of tablets for each formulation.
Friability
A tablet’s friability indicates the strength it is. The Roche Friabilator was utilized to examine the friability using the following procedure. In this test, many tablets have been thrown from a six-inch distance by a plastic chamber rotating at a speed of 25 rpm with each rotation, exposing them to the combined action of shock abrasion. The Roche friabilator was filled with a sample of six preweighed pills and turned on for 100 revolutions, or four minutes.
Percent friability (%F) was calculated as follows,
%Friability = (W1 – W2]*100/W1
Where, W1 = before test tablet weight
W2 = after test tablet weight
Weight variation test
Each of the 20 tablets in each formulation type was weighed individually with a weighing device in order to determine weight variance. Next, the weight of each tablet was averaged, and by comparing the weight of each tablet with the average, the weight deviation was found.
Disintegration time
The Pharmacopoeia’s standard tablet test was used to calculate the fast disintegrating tablet’s disintegration time. Following the insertion of the tablets in the disintegration tubes, the amount of time needed for the tablets to totally disintegrate without leaving any residue on the screen was measured to determine the disintegration time.
Time of Wetting
The amount of time as a tablet takes to dissolve on the tongue when it is held still is known as the “wetting time.” The hydrophilicity of the excipient and the internal structure of the tablets are strongly correlated with wetting time. A tissue paper piece that had been folded twice was put into a 10 cm-diameter petri plate that held 10 milliliters of water. After setting the tablet down on the paper, the number of seconds it took for it to fully wet was calculated. The process was slightly altered by keeping the water at 370C.
Chemical parameters of fast disintegrating tablet 13
Uniformity of drug content
1 ml of 0.1N HCL was used to dissolve the powder, which amounted to 10 mg of Loratadine, after the three tablets in each formulation had been weighed, crushed in a mortar, and powdered. Ten milliliters were diluted with 0.1N HCL after one milliliter of the stock solution was taken. Utilizing a double beam UV-visible spectrophotometer, the absorbance at 275 nm was measured. A formula has been employed to calculate content uniformity.
% Purity = Conc. of unknown/conc. of standard * 100
Dissolution study
Using the assist of a USP dissolving testing device type-Ⅱ (paddle type), the in vitro release of drugs was calculated. 900 milliliters of 0.1N HCL were used for the dissolution test, which was run at 75 rpm, 37 0C ± 0.5 0C. A 10-milliliter sample was taken at regular intervals of 3, 6, 9, 12, and 15 minutes from a zone that was at least 1 cm from the vessel wall and halfway between the dissolving medium’s surface and the revolving paddle’s top. To keep the volume of the medium constant, the volume that was removed was replaced with a new volume of dissolving media. Using 0.1N HCL as a blank, the filtered sample was subjected to spectrophotometric analysis at 275 nm. The drug release in the dissolution sample was calculated using the calibration curve.
Statistical analysis
The optimization of fast disintegrating tablets (FDT) was conducted by determining the optimal concentrations of two key factors: the superdisintegrant (crosspovidone) and the sublimating agent (camphor). This investigation utilized a central composite design approach. DESIGN EXPERT version 7.0.0 was used to analyze the obtained experimental data.
Effect of temperature and humidity
By studying the optimum batch placed in an environmental stability chamber maintained at 400C ± 20C or 75% ± 2% RH for 7, 14, 21, and 28 days, the effects of temperature and humidity were investigated. The tablet appearance, hardness, disintegration time, and medication content were all found to be constant.
Results and Discussion
Physical characterization of drug
Colour – White
Appearance – Fine powder
Taste – Bitter
Odour – Odourless
Determination of melting point
By using Thiele’s tube method, the melting point was determined to be 135.40C that complies with the reported literature.
Solubility
The solubility of Loratadine was studied in 0.1N HCL and distilled water. It found that Loratadine was soluble in distilled water at 0.0066 mg/ml and 0.1N HCL at 0.120 mg/ml.
λmax determination
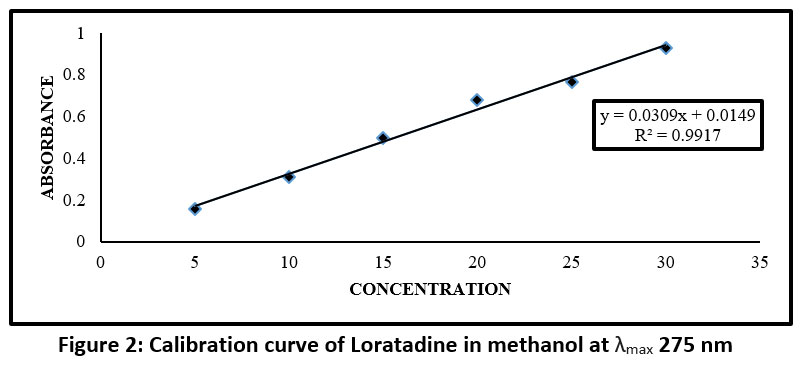
After the analysis of Loratadine in 0.1N HCL, the λ max was found to be 275 nm, which is accordance with the scientific literature.
Calibration curve in 0.1N HCL
Table 4: Data of absorbance for the Loratadine calibration curve in 0.1N HCL
| No. | Concentration (µg/ml) | Absorbance |
| a | 5 | 0.150 |
| b | 10 | 0.303 |
| c | 15 | 0.467 |
| d | 20 | 0.620 |
| e | 25 | 0.775 |
| f | 30 | 0.951 |
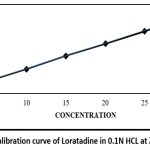 |
Figure 1: Calibration curve of Loratadine in 0.1N HCL at λmax 275 nmClick here to view Figure |
Calibration curve in methanol
Table 5: Absorbance values for calibration curve of Loratadine in methanol
| Sr no. | Concentration ppm (µg/ml) | Absorbance |
| a | 5 | 0.156 |
| b | 10 | 0.309 |
| c | 15 | 0.498 |
| d | 20 | 0.678 |
| e | 25 | 0.765 |
| f | 30 | 0.928 |
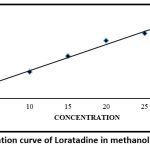 |
Figure 2: Calibration curve of Loratadine in methanol at λmax 275 nm Click here to view Figure |
IR studies
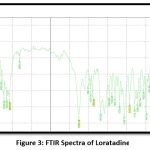 |
Figure 3: FTIR Spectra of LoratadineClick here to view Figure |
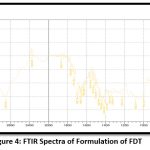 |
Figure 4: FTIR Spectra of Formulation of FDTClick here to view Figure |
The above FTIR spectrum showed the peaks of major functional groups of superdisintegrant and sublimating agent. When comparing the spectra of the drug mixture with the superdisintegrant, sublimating agent, and diluents, these peaks are nearly the same. Therefore, it can be concluded that the treatment, superdisintegrant, sublimating agent, and diluents that were employed in the formulation could not interact.
DSC studies
In Figure 5 DSC thermogram of Loratadine indicates a deep endotherm at 1360C, which is the drug’s melting point.In DSC thermogram of Loratadine and formulation the other weak endothermic peak at 1470C which is slightly shifted because presence of other excipients. Thus it confirms the presence of drug and other excipients in tablet formulation.
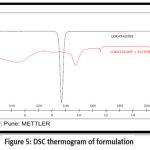 |
Figure 5: DSC thermogram of formulationClick here to view Figure |
Analysis of drug content in inclusion complexes:
All inclusion complexes had a drug content between 40 and 93%. This shows that the kneading procedure is effective and that the drug is loaded correctly into inclusion complexes. The drug composition of inclusion complexes is shown in Table 6.
Table 6: Percent drug content in inclusion complexes
| Sr no. | Stock solutionmedium | 1:1 inclusion complex drug content (%) | 1:2 inclusion complex drug content (%) |
| 1. | In distilled water | 41.9% | 80% |
| 2. | In 0.1N HCL | 67% | 93% |
Saturation solubility
The solubility of each inclusion complex was investigated in 0.1N HCL and pure water. The result of solubility of inclusion complexes prepared by kneading method as shown in Table 7. The dada indicated that solubility increased as increase in concentration of β-Cyclodextrin but highest increase of solubility was found in 1:2 inclusion complex as compared to 1:1 inclusion complex as shown in Fig 6.
Table 7: Solubility of Loratadine from various inclusion complexes
| Sr no. | Inclusion complexes | Solubility in pure water (mg/ml) | Solubility in 0.1N HCL (mg/ml) |
| 1. | 1:1 | 0.12 | 0.13 |
| 2. | 1:2 | 0.18 | 0.207 |
Dissolution studies
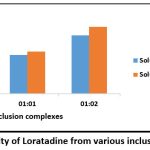 |
Figure 6: Solubility of Loratadine from various inclusion complexesClick here to view Figure |
The drug content analysis revealed that the 1:2 inclusion complex exhibited superior drug loading capabilities compared to the 1:1 ratio inclusion complex. Consequently, the 1:2 inclusion complex was chosen for further investigation in the dissolution study to assess drug release kinetics.
Table 8: Cumulative % drug (Loratadine) dissolved in distilled water
| Sr no. | Time (Min.) | % Drug release of Loratadine in pure water |
| 1 | 0 | 0 |
| 2 | 5 | 0.42% |
| 3 | 10 | 1.2% |
| 4 | 15 | 2.5% |
| 5 | 20 | 4.7% |
| 6 | 25 | 5.9% |
| 7 | 30 | 8.14% |
Table 9: Cumulative % drug (Loratadine) dissolved in 0.1N HCL
| Sr no. | Time (Min.) | % Drug release of Loratadine in 0.1N HCL |
| 1 | 0 | 0 |
| 2 | 5 | 40% |
| 3 | 10 | 42.2% |
| 4 | 15 | 44.5% |
| 5 | 20 | 46.5% |
| 6 | 25 | 48.7% |
| 7 | 30 | 50.19% |
Table 10: Cumulative % drug (Loratadine) dissolved from 1:2 inclusion complex prepared in pure water
| Sr no. | Time (Min.) | % Drug release from inclusion complex in pure wate |
| 1 | 0 | 0 |
| 2 | 5 | 54.85% |
| 3 | 10 | 58.28% |
| 4 | 15 | 65.57% |
| 5 | 20 | 71.14% |
| 6 | 25 | 76.23% |
| 7 | 30 | 79.65% |
Table 11: Cumulative % drug (Loratadine) dissolved from 1:2 inclusion complex prepared in 0.1N HCL
| Sr no. | Time (Min.) | % Drug release from inclusion complex in 0.1N HCL |
| 1 | 0 | 0 |
| 2 | 5 | 77.22% |
| 3 | 10 | 79.74% |
| 4 | 15 | 82.35% |
| 5 | 20 | 85.41% |
| 6 | 25 | 88.83% |
| 7 | 30 | 92.43% |
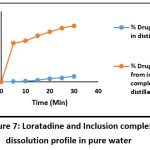 |
Figure 7: Loratadine and Inclusion complex dissolution profile in pure waterClick here to view Figure |
Evaluation of Pre-compression properties of the drug and its excipients
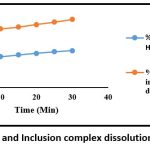 |
Figure 8: Loratadine and Inclusion complex dissolution profile in 0.1N HCLClick here to view Figure |
The powder mixtures that were prepared were examined for blend properties, including hausner’s ratio, tapped density, bulk density, and angle of repose compressibility index. The results obtained are displayed in Table 12.
Table 12: Physical parameters of the powder blend
| Parameters Batches | Angle of repose | Bulk density(g/ml) | Tapped density(g/ml) | Compressibility index(%) | Hausner’s ratio |
| B1 | 35.53 | 0.6 | 0.7 | 14.28 | 1.16 |
| B2 | 32 | 0.6 | 0.7 | 14.31 | 1.16 |
| B3 | 29.05 | 0.6 | 0.7 | 13.28 | 1.25 |
| B4 | 29.74 | 0.6 | 0.7 | 13.26 | 1.27 |
| B5 | 34.59 | 0.6 | 0.7 | 13.28 | 1.16 |
| B6 | 32.82 | 0.6 | 0.7 | 15.10 | 1.17 |
| B7 | 32 | 0.6 | 0.7 | 14.56 | 1.16 |
| B8 | 32 | 0.6 | 0.7 | 15.01 | 1.17 |
| B9 | 32 | 0.6 | 0.7 | 13.02 | 1.16 |
| B10 | 32 | 0.6 | 0.7 | 13.02 | 1.17 |
| B11 | 32 | 0.6 | 0.7 | 13.01 | 1.25 |
| B12 | 32 | 0.6 | 0.7 | 13.02 | 1.18 |
| B13 | 32 | 0.6 | 0.7 | 13.01 | 1.16 |
According to all of these results, the powder blend complies with acceptable limits and showed adequate compressibility and flow properties into the die cavity.
Physical Parameters of Fast Disintegrating Tablet
The tablets passed physical evaluation based on characteristics including weight fluctuation, hardness, thickness, and friability, as well as chemical parameters like wetting time, disintegration time, uniformity drug content, and dissolution study. Every batch fulfilled the required weight variation, hardness, thickness, and friability, DT, wetting time, uniformity of drug content, and drug release parameters, as well as other acceptable standards, and showed adequate mechanical strength.
Table 13: Physical parameters of fast disintegrating tablets
| Parameters Batches | Weight variation | Thickness(mm) | Hardness(kg/cm2) | Friability(%) |
| B1 | 149.8 ± 0.43 | 3.26 ± 0.04 | 3.11 ± 0.20 | 0.3 ± 0.14 |
| B2 | 150.15 ± 0.32 | 3.2 ± 0.07 | 2.93 ± 0.35 | 0.1 ± 0.63 |
| B3 | 149.95 ± 0.50 | 3.25 ± 0.07 | 2.8 ± 0.31 | 0.2 ± 0.66 |
| B4 | 150.15 ± 0.32 | 3.37 ± 0.04 | 2.88 ± 0.28 | 0.2 ± 0.52 |
| B5 | 149.65 ± 0.66 | 3.3 ± 0.04 | 2.9 ± 0.26 | 0.3 ± 0.52 |
| B6 | 149.8 ± 0.43 | 3.36 ± 0.04 | 3.03 ± 0.22 | 0.3 ± 0.14 |
| B7 | 149.9 ± 0.14 | 3.26 ± 0.05 | 3 ± 0.28 | 0.2 ± 0.66 |
| B8 | 149.65 ± 0.66 | 3.23 ± 0.09 | 3.05 ± 0.23 | 0.1 ± 0.63 |
| B9 | 150.15 ± 0.32 | 3.24 ± 0.05 | 3.01 ± 0.21 | 0.2 ± 0.66 |
| B10 | 149.8 ± 0.43 | 3.37 ± 0.04 | 2.93 ± 0.24 | 0.3 ± 0.14 |
| B11 | 149.9 ± 0.50 | 3.3 ± 0.07 | 2.85 ± 0.26 | 0.3 ± 0.52 |
| B12 | 149.65 ± 0.66 | 3.26 ± 0.05 | 3.1 ± 0.23 | 0.2 ± 0.52 |
| B13 | 149.8 ± 0.43 | 3.24 ± 0.05 | 3.11 ± 0.12 | 0.3 ± 0.14 |
Table 14: Results of disintegration time, wetting time, and uniformity of content of Loratadine Fast Disintegration Tablet.
| Parameters Batches | Disintegration time (sec) | Time of Wetting (sec) | Uniformity of content(%) |
| B1 | 49.33 ± 0.02 | 52.26 ± 0.08 | 75.2 ± 0.7 |
| B2 | 39.15 ± 0.019 | 50.17 ± 0.02 | 82.6 ± 0.4 |
| B3 | 41.36 ± 0.06 | 51.27 ± 0.01 | 78.4 ± 0.8 |
| B4 | 20.22 ± 0.04 | 39.24 ± 0.02 | 102.9 ± 0.2 |
| B5 | 51.25 ± 0.03 | 57.17 ± 0.02 | 72.25 ± 1 |
| B6 | 26.19 ± 0.02 | 41.26 ± 0.01 | 96.7 ± 0.2 |
| B7 | 35.24 ± 0.03 | 45.37 ± 0.01 | 91.05 ± 1.2 |
| B8 | 43.37 ± 0.3 | 56.14 ± 0.02 | 85.7 ± 0.3 |
| B9 | 32.21 ± 0.03 | 48.32 ± 0.01 | 92.95 ± 0.3 |
| B10 | 32.30 ± 0.02 | 48.21 ± 0.01 | 92.9 ± 0.3 |
| B11 | 32.20 ± 0.03 | 48.30 ± 0.01 | 92.82 ± 0.2 |
| B12 | 32.21 ± 0.03 | 48.33 ± 0.01 | 92.96 ± 0.3 |
| B13 | 32.21 ± 0.03 | 48.30 ± 0.01 | 92.81 ± 0.3 |
Chemical parameters of fast disintegrating tablet
In – vitro drug dissolution study
USP dissolving test apparatus was used to perform in vitro drug dissolution experiments in 0.1N HCL.
To ascertain whether the formulation of the fast disintegrating tablets increased the availability of Loratadine. After calibrating the temperature to 370C ± 0.50C at 75 rpm, After 3, 6, 9, 12, 15, and 18 minutes, the samples were taken out. The results are listed below.
Table 15. Result of % drug release of all formulations
| Parameter Batches | % Cumulative drug release |
| B1 | 72.94 ± 0.08 |
| B2 | 84.25 ± 0.58 |
| B3 | 80 ± 0.53 |
| B4 | 101.6 ± 0.22 |
| B5 | 68.85 ± 0.92 |
| B6 | 98.2 ± 0.14 |
| B7 | 91.8 ± 0 |
| B8 | 83.7 ± 0.08 |
| B9 | 97.8 ± 0.43 |
| B10 | 97.8 ± 0.4 |
| B11 | 97.1 ± 0.4 |
| B12 | 97.8 ± 0.43 |
| B13 | 97.6 ± 0.42 |
Statistical analysis
Central Composite Design
The amount of the superdisintegrant (crospovidone X1) and sublimating agent (camphor X2) was chosen as the independent variable in a 22 central composite design. A statistical model including polynomial and interaction components was used to evaluate the responses. The interactions and effects of the constituents were represented by the coefficients of a polynomial equation that was generated.
Table 16: Results of 22 Central Composite Design: Effects of two factors on both the responses
| No. | Crospovidone(%) | Camphor(%) | Responses | |
| DT (sec) | %CDR | |||
| 1 | 5 | 10 | 49.33 | 72.94 |
| 2 | 10 | 10 | 39.15 | 84.25 |
| 3 | 5 | 15 | 41.36 | 80 |
| 4 | 10 | 15 | 20.22 | 101.6 |
| 5 | 3.96 | 12.50 | 51.25 | 68.85 |
| 6 | 11.04 | 12.50 | 26.19 | 98.2 |
| 7 | 7.50 | 8.96 | 35.24 | 91.8 |
| 8 | 7.50 | 16.04 | 43.37 | 83.7 |
| 9 | 7.50 | 12.50 | 32.21 | 97.8 |
| 10 | 7.50 | 12.50 | 32.30 | 97.8 |
| 11 | 7.50 | 12.50 | 32.20 | 97.1 |
| 12 | 7.50 | 12.50 | 32.21 | 97.8 |
| 13 | 7.50 | 12.50 | 32.21 | 97.6 |
Full model for A (Disintegration time)
Disintegration time = 35.94 – 8.35A – 1.93B
It became apparent that the superdisintegrant, crospovidone, had a favorable effect on the disintegration time.
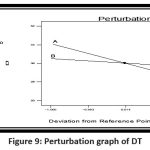 |
Figure 9: Perturbation graph of DTClick here to view Figure |
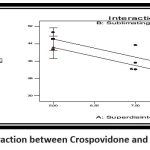 |
Figure 10: Interaction between Crospovidone and Camphor in DTClick here to view Figure |
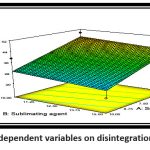 |
Figure 11: Effect of independent variables on disintegration time 3D surface plotClick here to view figure |
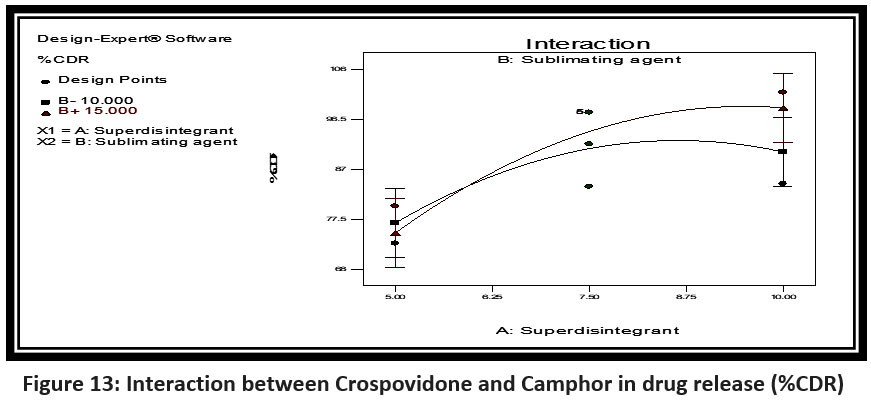
Full model for B (%Drug release)
Drug release = 97.80 – 9.30A + 1.62B – 7.37A2 – 5.6B2 + 2.57AB
It was observed that the superdisintegrant (crosspovidone) and sublimating agent (camphor) had a positive effect on drug release.
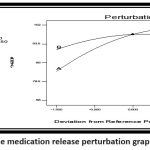 |
Figure 12: The medication release perturbation graph (%CDR)Click here to view Figure |
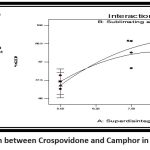 |
Figure 13: Interaction between Crospovidone and Camphor in drug release (%CDR)Click here to view Figure |
 |
Figure 14: Effect of independent variables on the 3D surface plot of drug release (%CDR)Click here to view Figure |
Table 17: Optimized concentration obtained on the basis of DT and drug release
| Sr no. | Factors | Optimized amount (%) |
| 1 | Superdisintegrant | 9.22 |
| 2 | Sublimating agent | 13.31 |
Final formulation was prepared by using the above concentration and tablets were evaluated for various parameters.
Evaluation parameters of optimized batch
Table 18: Evaluation parameters of optimized batch (F14)
| Sr no. | Parameters | Results |
| 1 | Angle of repose | 29.74 (Excellent) |
| 2 | Density of bulk (g/ml) | 0.6 |
| 3 | Density with a tap (g/ml) | 0.7 |
| 4 | Index of Compressibility (%) | 14.28 (Good) |
| 5 | Hausner’s ratio | 1.16 (Good) |
| 6 | Weight variation | 150.1 ± 0.46 |
| 7 | Thickness (mm) | 3.26 ± 0.04 |
| 8 | Hardness (kg/cm3) | 3.11 ± 0.2 |
| 9 | Friability (%) | 0.3 ± 0.78 |
| 10 | Time of wetting | 30.67 ± 0.42 |
| 11 | Uniformity of content (%) | 100.63 ± 0.82 |
| 12 | Disintegration time | 29.58 ± 0.44 |
| 13 | % Drug release | 101.25 ± 0.45 |
Effect of temperature and humidity
Table 19: Effect of temperature and humidity on optimized batch
| Specification | Days | ||||
| zero | Seven | Fourteen | Twenty One | Twenty Eight | |
| Hardness(kg/cm2) | 3.11 ± 0.05 | 3.1 ± 0.05 | 3.1 ± 0.05 | 3.11 ± 0.005 | 3.10 ± 0.05 |
| DT (sec) | 29.58 ± 0.04 | 29.51 ± 0.04 | 29.58 ± 0.04 | 28.49 ± 0.04 | 29.51 ± 0.04 |
| Dug content (%) | 100.63± 0.82 | 100.61± 0.82 | 100.5 ± 0.82 | 100.56± 0.82 | 100.41± 0.82 |
From The above-mentioned results indicate that there were no major physical or chemical changes in the optimized batch after one month.
Conclusion
The findings suggest that solubility enhancement was achieved through complexation. Overall, formulation B14, containing 9.22% crospovidone and 13.31% camphor, emerged as the superior candidate, meeting all criteria for a fast-disintegrating tablet. Enhanced dissolution of Loratadine may translate to improved bioavailability, efficacy, and ultimately, enhanced patient compliance.
Acknowledgement
Authors thanks to Glenmark Pharmaceutical Pvt. Ltd, Taloja, Mumbai for providing gift sample of Loratadine. The authors are also thankful to METs institute of Pharmacy, Nashik for their facilities in carrying out this research work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Authors’ Contribution
Dr. D.S. Bhambere : oversaw the work,
Ms. Samiksha Amrutkar : conducted all the tests.
Mr. Shivam K. Navale, Ms. Pooja Birari, Ms. Shweta Shevatkar, Dr. Sapana P. Ahirrao and Dr. Sanjay J. Kshirsagar : prepared the manuscript
References
- AlMasoud, Najla, “Loratadine.” Profiles of Drug Substances, Excipients and Related Methodology. 47 (2022): 55-90. https://www.go.drugbank.com/drugs/DB00455
CrossRef - Chavda H, Patel C, Anand I. Biopharmaceutics classification system. Systematic reviews in pharmacy. 2010; 1(1):62.
CrossRef - Patrick J. Sinko., Martin’s Physical Pharmacy and Pharmaceutical sciences, Sixth edition, 2006; 205
- Agarwal V, Bhavesh H, Kothari, Derek V. Moe R K. Khankari. Drug Delivery: Fast-Dissolve Systems. In: James swarbrick. Encyclopedia of pharmaceutical technology, 3rded.USA: Informa healthcare 2007:1104-5.
- Nangude TD, Chatap .VK, Bhise KS, Sharma D.K. Mouth dissolving tablets: Geriatrics and pediatrics friendly drug delivery system. Indian Drugs 2007; 44(6): 471-3.
- Indurwade NH, Rajyaguru TH, Nakhat PD. Novel approach- Fast dissolving tablets. Indian Drugs 2002; 39(8): 405.
- Pharmacopoeia I. Indian pharmacopoeia. Ghaziabad: Indian Pharmacopoeia Commission. 2007:1516-7.
- Maragos S, Archontaki H, Macheras P, Valsami G. Effect of cyclodextrin complexation on the aqueous solubility and solubility/dose ratio of praziquantel. AAPS PharmSciTech. 2009;10:1444-51.
CrossRef - Fernandez-Lopez S, Kim HS, Choi EC, Delgado M, Granja JR, Khasanov A, Kraehenbuehl K, Long G, Weinberger DA, Wilcoxen KM, Ghadiri MR. Antibacterial agents based on the cyclic D, L-α-peptide architecture. Nature. 2001 26; 412(6845):452-5.
CrossRef - Patel R, Bhimani D, Patel J, Patel D. Solid-state characterization and dissolution properties of ezetimibe–cyclodextrins inclusion complexes. Journal of Inclusion Phenomena and Macrocyclic Chemistry. 2008;60: 241-51.
CrossRef - Ghodke DS, Nakhat PD, Yeole PG, Naikwade NS, Magdum CS, Shah RR. Preparationa and Characterization of domperidone Inclusion complexes with cyclodextrin: Influence of preparation method. Iranian journal of pharmaceutical research. 2009 31; 8(3):145-51.
- Gholve S, Todkar G, Bhusnure O, Jadhav A, Rajurkar R, Thonte S. Formulation And Evaluation of Fast Dissolving Tablets of Etoricoxib. World Journal of Pharmacy and Pharmaceutical Sciences. 2015 3;4(10):1357-76.
- Vaghela B, Kayastha R, Bhatt N, Pathak N, Rathod D. Development and validation of dissolution procedures. Journal of applied pharmaceutics.

This work is licensed under a Creative Commons Attribution 4.0 International License.




{kind=link}
{kind=link}