
Design, Synthesis and Molecular Modeling Study of Quinazolin-4(3H) Ones With Potential Anti-Inflammatory Activity
Pharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Zagazig University, Zagazig Egypt.
Download this article as:
![]()
The present investigation is concerned with the synthesis of different 2-methyl-4(3H)-quinazolinones with the objective of discovering novel and potent anti-inflammatory agents. Structures of the synthesized compounds were elucidated by spectral and elemental analyses. Some of the obtained compounds were evaluated for their anti-inflammatory activity as well as gastric ulcerogenic effects. Results showed that 2-[2-methyl-4-oxo-3(H)-quinazolin-3-yl]-N-(4-chlorophenyl-2-thiaoxothiazol-3-yl) acetamide (11 b) showed higher anti-inflammatory activity in carageenan-induced rat paw edema test with low gastric ulcerogenicity compared with indomethacin. Furthermore, molecular modeling studies were performed in order to rationalize the obtained biological results.
KEYWORDS:4(3H)Quinazolinone; anti-inflammatory; docking; COX-2
Introduction
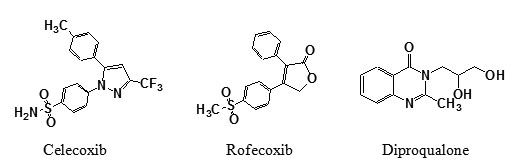
Non-steroidal anti-inflammatory drugs (NSAIDs), which are widely used for reducing pain and swelling associated with inflammation, represent a research area of continuous and ever-growing development. It was discovered that traditional NSAIDs show very little selectivity for COX-2, the fact that prompted to suggest [1] that the therapeutic anti-inflammatory action of NSAIDs is produced by inhibition of COX-2, while the unwanted side effects arise from inhibition of COX-1 activity. Accordingly, a great deal of interest has been devoted in recent years to the discovery of selective COX-2 inhibitors. As a result of these investigations, the two first COX-2 selective inhibitors: celecoxib [2] and rofecoxib [3] are already in clinical use. It was established that quinazolinones have powerful anti-inflammatory activity. Diproqualone 1 is well known drug which used primarily for the treatment of inflammatory pain associated with osteoarthritis [4].Furthermore, it was reported that many compounds having a 1,3,4-oxadiazole [5,6], 1,3,4-thiadiazole [7,8], 1,2,4-triazole [9,10] skeleton possessed significant anti-inflammatory activity with no gastrointestinal toxicity. On the basis of the above mentioned reports, the present investigation is concerned with the synthesis of different 2-methyl-3-substituted 4-(3H)- quinazolinone derivatives with the objective of discovering novel and potent anti-inflammatory agents that might be devoid of the gastrointestinal side effects.
Results and discussion
Chemistry
Synthesis of the title compounds 5a,5b,6,7,8 and 9 has been carried out as depicted in Scheme 1. Anthranilic acid (1) was refluxed in acetic anhydride to generate the previously reported benzoxazin-4-one (2) [11] which underwent cyclization by treatment with ethyl glycinate HCl to form the ester derivative (3) [12]. The treatment of (3) with hydrazine hydrate resulted in the formation of the acid hydrazide (4) [13].The key intermediate (4) was reacted with cyclic anhydride namely succinic and phythalic anhydride to give succinamido and phythalimido derivatives (5 a, b) respectively. Also, compound (4) was reacted with p-toluene sulphonyl chloride to give the sulphonamide (6). Reaction of the acid hydrazide (4) with 5,5-dimethyl-1,3-cyclohexanedione (dimedone) in toluene gave the enaminone derivative (7) which was reacted with 3-p-nitrophenyl-2-cyano acrylonitrile in ethanol under reflux to give the hexahydroquinoline derivative (8). compound (4) underwent cyclization when reacted with ethyl chloroformate in n-butanol under reflux to give 5-oxo-1,3,4-oxadiazole (9).
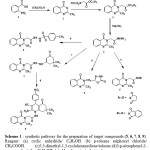 |
Scheme 1 : synthetic pathway for the preparation of target compounds (5, 6, 7, 8, 9). Reagent: (a) cyclic anhydride/ C2H5OH. (b) p-toluene sulphonyl chloride/ CH3COOH. (c)5,5-dimethyl-1,3-cyclohexanedione/toluene.(d)3-p-nitrophenyl-2-cyanoacrylonitrile /C2H5OH. (e) chloroformate / n-butanol. |
the acid hydrazide (4) was reacted with carbon disulphide in ethanolic potassium hydroxide to give the potassium dithiocarbazate salt (10) which was cyclized into 1,3-thiazoles (11a-c), 1,3,4-oxadiazole (12)[14], 1,3,4-thiadiazole (13) and 1,2,4-triazole (14) derivatives via the reaction with phenacyl bromides derivatives, reflux in ethanol, stirring with Conc. H2SO4 and reaction with hydrazine hydrate, respectively, furthermore, the oxadiazole (12) and thiadiazole (13) were alkylated via the reaction with different alkyl halides in DMF containing potassium carbonate to give the thioether derivatives(15),(16) (Scheme 2).
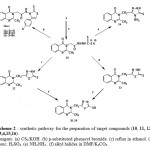 |
Scheme 2 : synthetic pathway for the preparation of target compounds (10, 11, 12 , 13, 14,15,16). Reagent: (a) CS2/KOH. (b) p-substituted phenacyl bromide. (c) reflux in ethanol. (d) Conc. H2SO4. (e) NH2NH2 . (f) alkyl halides in DMF/K2CO3. |
4-Amino, 1,2,4-triazole-5-thione derivative (14), was reacted with aromatic aldehyde and phenyl isothiocynate to give azomethine derivatives (17a,b) and thiourea derivative (18) respectively. The triazole (14) was cyclized into the triazolothiazole derivatives (19), (20) and the triazolothiadiazine derivative (21) by the reaction with acetic acid in phosphorous oxychloride, carbon disulphide and p-chlorophenacyl bromide, respectively (Scheme 3).
Scheme
synthetic pathway for the preparation of target compounds (5, 6, 7, 8, 9). Reagent: (a) cyclic anhydride/ C2H5OH. (b) p-toluene sulphonyl chloride/ CH3COOH. (c)5,5-dimethyl-1,3-cyclohexanedione/toluene.(d)3-p-nitrophenyl-2-cyanoacrylonitrile /C2H5OH. (e) chloroformate / n-butanol.
Scheme
synthetic pathway for the preparation of target compounds (10, 11, 12 , 13, 14,15,16). Reagent: (a) CS2/KOH. (b) p-substituted phenacyl bromide. (c) reflux in ethanol. (d) Conc. H2SO4. (e) NH2NH2 . (f) alkyl halides in DMF/K2CO3.
| Comp. No | Y | R | Comp.
No |
Y | R |
| 15a | O | CH3
|
15f | O | CH2CONH-(o-OCH3)C6H4 |
| 15b | O | CH2CH3
|
16a | S | CH3
|
| 15c | O | CH2C6H5 | 16b | S | CH2CONH-C6H5 |
| 15d | O | CH2CONH-C6H5 | 16c | S | CH2CONH-(p-Cl)C6H4 |
| 15e | O | CH2CONH-(m-OCH3)C6H4 | 16d | S | CH2CONH-(p-OCH3)C6H4 |
Scheme3
synthetic pathway for the preparation of target compounds (17,18,19,20,21). Reagent: (a) ArCHO/C2H5OH. (b) PhNCS / DMF. (c) CH3COOH /POCl3. (d) CS2/KOH. (e) p-chloro phenacyl bromide.
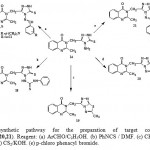 |
Scheme 3: synthetic pathway for the preparation of target compounds (17,18,19,20,21). Reagent: (a) ArCHO/C2H5OH. (b) PhNCS / DMF. (c) CH3COOH /POCl3. (d) CS2/KOH. (e) p-chloro phenacyl bromide. |
Biological evaluation
The in vivo evaluation of anti-inflammatory activity of the synthesized compounds (11a, 11b, 15c,15d, 16 a, 16b, 14, 20) was accomplished using carrageenin-induced rat paw edema method in comparison with celecoxib as a standard using the method by Winter et al[15].
Results of the anti-inflammatory activity of the tested compounds as well as celecoxib are shown in Table 1. Results showed that most of the tested compounds exhibited significant (P < 0.05) inhibition against carrageenan-induced rat paw edema and comparable anti-inflammatory activity relative to celecoxib. Among these derivatives, compounds 11b and 15c were found to be more potent than celecoxib.
Gastric ulceration
Gastric ulcerogenic effects were determined in rats [16] for representative examples of the synthesized compounds, (11a, 11b, 15c,15d, 16 a, 16b, 14, 20). Results indicated that the tested compounds exhibited low gastric ulcerogenicity compared with indomethacin. Table 2.
Table 1: The anti-inflammatory activity of the tested compounds and celecoxib at dose of 0.9 mg/100 gm body weight of rats on the inflamed rat paw (n=5 rats.).
| G
Groups |
Thickness of rat paw (mm) | |||
| 1 hour | 2 hours | 3 hours | 4 hours | |
|
Control |
0.77± 0.025a |
0.86± 0.037a |
0.87± 0.032a |
0.88 ±0.025a |
|
Celecoxib |
0.42 ±0.025c |
0.44± 0.024def |
0.45 ±0.029de |
0.49± 0.013de |
|
11a |
0.60± 0.041b |
0.61± 0.031bc |
0.60± 0.041bc |
0.65± 0.029bc |
|
11b |
0.32± 0.025c |
0.35± 0.020f |
0.37±0.025e |
0.37± 0.025f |
|
15c |
0.42± 0.047c |
0.42± 0.031ef |
0.42± 0.025e |
0.43± 0.024ef |
| 15d | 0.62± 0.047b | 0.65± 0.028b | 0.67± 0.047b | 0.67± 0.047bc |
|
16a |
0.65± 0.086b |
0.70± 0.057b |
0.71± 0.059b |
0.72± 0.047b |
|
16b |
0.40± 0.041c |
0.42± 0.031ef |
0.46± 0.037 de |
0.41± 0.031ef |
|
14 |
0.46± 0.023c |
0.51± 0.012cd |
0.55± 0.028cd |
0.57± 0.025cd |
|
20 |
0.43± 0.034c |
0.52± 0.047cd |
0.60± 0.041bc |
0.60± 0.041c |
Means are expressed as Mean ± S.E
Means within the same column carrying different litters are significant at (P≤0.05).
Table 2 : ulcerogenic activity of the tested compounds and indomethacin (n=5 rats). (mean± S.E.).
| Group
|
Incidence of gastric ulcer | Mean
ulcer score |
Ulcer
index |
| Control | 0.0 % | 0.0 | 0.0 |
| Indomethacin | 100 % | 4±0.035 | 500 |
| 11 a | 60 % | 2±0.050 | 120 |
| 11 b | 60 % | 2±0.035 | 120 |
| 15 c | 20 % | 1±0.080 | 20 |
| 15 d | 80 % | 3±0.080 | 240 |
| 16 a | 80 % | 3±0.090 | 240 |
| 16 b | 20 % | 1±0.025 | 20 |
| 14 | 20 % | 1±0.070 | 20 |
| 20 | 60 % | 2±0.027 | 120 |
Statistical analysis
Data were analyzed using computer program SPSS.
Molecular modeling study
Molecular dockings studies of compounds(11b, 15c,15d, 16 a, 16b, 14, 20) was performed in order to rationalize the obtained biological results. Besides, molecular docking studies helped in understanding the various interactions between the ligands and enzyme active site in details. Docking studies of the inhibitors were performed by MOE (Molecular Operating Environment) using murine cyclooxygenase-2 co-crystallized with selective inhibitor, celecoxib (Protein Data Bank code PDB ID: 6COX) as a template. We performed 100 docking iterations for each ligand and the top-scoring configuration of the ligand-enzyme complexes was selected on energetic grounds.
1- Docking of (11b) revealed several molecular interactions considered to be responsible for the observed affinity including three hydrogen bonds between the ligand carbonyl group and His 90 (distance 2.71 A°), the OH group of Tyr355 (distance 2.93 A°) and Arg 513 (distance 3.45 A°). Furthermore, the ligand p-chlorophenyl moiety was located in the vicinity of the aromatic pocket forming hydrophobic interactions with the side chains of Val 116, Val 349, Val 523, Phe 518, Leu395, Leu 531 and Met 113. (fig.1)
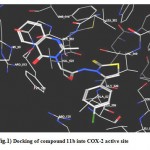 |
Figure 1: Docking of compound 11b into COX-2 active site.
|
2- Docking of (15 c) revealed two types of molecular interactions considered to be responsible for the observed affinity including A hydrogen bond interaction between the ligand carbonyl group and His 90 (distance 2.60 A°) and another hydrophobic interaction between the ligand benzyl moiety and the hydrophobic pocket formed by the side chains of Val 349, Val 523, Phe 518, Leu352,and Trp 387. (fig.2)
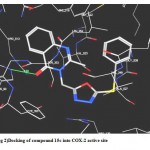 |
Figure 2: Docking of compound 15c into COX-2 active site.
|
3- Docking of (15 d) showed that the ligand was oriented so that the phenyl moiety fill the hydrophobic pocket formed by the side chains of Val 349, Val 523, Val 344, Phe 205,Phe 518, Leu352,and Leu 531. Also it showed three hydrogen bonds between the ligand carbonyl group His 90 (distance 2.24 A°), the OH group of Tyr355 (distance 3.29 A°) and Arg 513 (distance 3.43 A°).(fig 3)
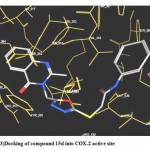 |
Figure 3: Docking of compound 15d into COX-2 active site.
|
4- Docking of (16 a) showed A hydrogen bond interaction between the ligand carbonyl group and Ser 353 (distance 3.34 A°), and a hydrophobic interaction between the ligand and the hydrophobic pocket side chain of Val 523.(fig.4)
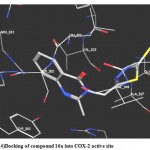 |
Figure 4:Docking of compound 16a into COX-2 active site.
|
5- Docking of (16 b) revealed that the carbonyl group in the side chain of the ligand has two hydrogen bonds interaction with His 90 (distance 2.72 A°) and and Keu 352 (distance 2.82 A°). In addition to a hydrophobic interaction between the ligand phenyl moiety and the hydrophobic pocket formed by the side chains of Val 523, Phe 518 and Ile 517.(fig. 5)
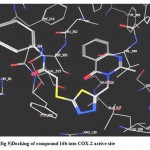 |
Figure 5:Docking of compound 16b into COX-2 active site.
|
6- Docking of (14) revealed several molecular interactions including a hydrogen bond between the ligand carbonyl group and Ser 353 (distance 3.04 A°), and another two hydrogen bonds between the ligand amino group and Gln 192 (distance 3.18 A°) and Leu 352 (distance 2.44 A°). The ligand showed also a hydrophobic interaction with the hydrophobic pocket formed by the side chains of Val 349, Leu35 and Leu 53.(fig.6)
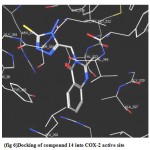 |
Figure 6: Docking of compound 14 into COX-2 active site.
|
7-Docking of (20) revealed two types of molecular interactions considered to be responsible for the observed affinity including a hydrogen bond interaction between the ligand carbonyl group and His 90 (distance 3.38 A°) and a hydrophobic interaction between the ligand and the hydrophobic pocket side chain of Val 523, Phe 518 and Ile 517.(fig. 7)
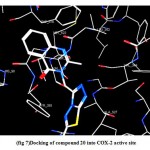 |
Figure 7: Docking of compound 20 into COX-2 active site.
|
Conclusion
We reported here the synthesis of different 2-methyl-3-substituted 4-(3H)- quinazolinone derivatives. The synthesized compounds (11a, 11b, 15c,15d, 16 a, 16b, 14, 20) were tested for their anti-inflammatory activity. Results showed that most of the tested compounds exhibited significant anti-inflammatory activity in the carageenan-induced rat paw edema test with low gastric ulcerogenicity compared with indomethacin. Furthermore, the molecular modeling studies suggested that 1,3-thiazole derivative (11b) exhibited higher affinity for the COX-2 active site and hence increased anti-inflammatory activity than the rest of the tested compounds.
Experimental
Chemistry
Melting points were determined using Gallen Kamp digital melting point apparatus and were uncorrected. IR spectra were recorded as KBr disk using Shimadzu IR FTIR 8000 spectrophotometer. 1H NMR spectra were carried out on Varian Gemini 200, 200 MHz, using DMSO-d6 or CDCl3 as a solvent (the chemical shifts are given in δ, ppm). All NH and NH2 protons were exchangeable with D2O. Mass spectrum was performed on Joel, JMS-600 spectrometer at an ionization voltage of 70 eV (Joel, Tokyo, Japan). Elemental analyses were performed at microannalytical center, Faculty of Science Cairo University, Giza, Egypt. All reactions were monitored by thin-layer chromatography (TLC) using silica gel 60 GF245 precoated sheets 20 × 20 cm, layer thickness 0.2 mm (E-Merck, Germany) and were visualized by UV-lamp at wave length (λ) 254 nm.
Compounds 1, 2,3,4,11 were prepared according to reported procedures [11].[12], [13], [14].
Synthesis of 2-(2-Methyl-4-oxo-3(H)-quinazolin-3-yl)-N-substituted acetamides (5a,b).
A mixture of (4) (0.01 mol.) and appropriate acid anhydride namely, succinic unhydride and/or phthalic unhydride (0.01 mol.) in glacial acetic acid (20 ml) was stirred under reflux for 8hours. After cooling the obtained precipitate was filtered, washed with water and crystallized from absolute ethanol.
(5a) Yield 39%; mp 211-212 °C. IR (KBr, cm): 3300 ( NH ), 3030( CH Ar ), 2944 ( Aliph. ), 1793, 1735, 1708, 1680 ( 4C=O ). 1HN M R(200 MHz, DMSO-d6, d ppm): 2.39, 2.41 (t, 4H ), 2.7 (s, 3H, CH3 ), 5.04 ( s, 2H, CH2 ), 7.48-8.14 ( m, 4H, ArH ), 11.05 ( s, 1H, NH exchangeable). Anal. calcd for (C15H14N4O4) C 57.32, H 4.49, N 17.83; found: C 56.93, H 4.59, N 17.83.
(5b) Yield 60%; mp 305-307 °C. IR (KBr, cm): 3105 ( NH ), 3020 ( CH Ar ), 1798, 1745, 1708, 1680 ( 4C=O ). 1HN M R(200 MHz, DMSO-d6, d ppm): 2.59 ( s, 3H, CH3 ), 5.11 ( s, 2H, CH2 ), 7.48-8.15 ( m, 8H, ArH ), 11.36 ( s, 1H, NH exchangeable). Anal. calcd for (C19H14N4O4) C 62.98, H 3.89, N 15.46; found: C 62.49, H 4.49, N 15.17..
Synthesis of 2-(2-Methyl-4-oxo-quinazolin-3-yl)N-([(4-tolyl)sulphonyl] acetohydrazide(6). To a solution of the acid hydrazide (4) (0.01 mol.) in glacial acetic acid (20 ml), p-toluene sulphonyl chloride (0.01 mol.) was added. The reaction mixture was stirred for 12 hours at room temperature, water was added and the precipitate obtained was filtered off and crystallized from absolute ethanol. Yield 74%, m.p. 245-246 °C. IR (KBr) cm-1: 3301, 3170 ( 2 NH ), 3061 ( CH Ar. ), 2964, 2929 ( CH Aliph. ), 1706, 1681 ( 2 C=O ), 1339,1162 ( SO2 ). 1HMNR DMSO- d6 (δ ppm): 2.32, 2.41 (s, 6H, 2CH3), 4.7 (s, 2H, NCH2), 7.30-8.10 (m, 8H, ArH), 9.97, 10.60 (s, 2H, 2NH exchangeable). Anal. calcd for (C18H18N4O4S) C 55.95 , H 4.70, N 14.50; found: C 55.80, H 5.07, N 15.02.
Synthesis of N’-(5, 5-Dimethyl-3-oxo-cyclohex-1-enyl)-2-(4-oxo (3H)-2-methyl-quinazolin-3-yl) acetohydrazide (7 ).
A mixture of 5, 5-dimethyl-1, 3-cyclohexanedione (dimedone) (0.01 mol.) and Acid hydrazide (4) (0.01 mol.) in toluene was heated under reflux for 15 hours, the reaction mixture was allowed to cool at room temperature, the obtained solid product was filtered off, dried and crystallized from absolute ethanol. Yield 91%, m.p. 260-261°C. IR (KBr) cm-1: 3221 ( 2 NH ), 3032 ( CH Ar ), 2952 ( CH Aliph.), 1677 ( 3C=O ). 1HNMR DMSO-d6 (δ ppm): 0.99 ( s, 3H, CH3 ), 1.05 ( s, 3H, CH3 ), 2.1 ( s, 2H, CH2 ), 2.29 ( s, 2H, CH2 ), 2.6 ( s, 3H, CH3 ), 4.99 ( s, 2H, NCH2), 5.2 ( s, 1H, =CH ), 7.4-8.12 ( m, 4H, Ar-H ), 8.8 ( s, 1H, =CNH exchangeable),10.3 (s, 1H, NHCO exchangeable). Ana. calcd for : (C19H22N4O3) C 64.39, H 6.26, N 15.81; found: C 64.58, H 6.33, N 15.60.
Synthesis of N-(2-Amino-4-p-nitro phenyl- 7, 7-dimethyl- 1, 4, 5, 6, 7, 8-hexahydro-3-cyano-5-oxo-quinolin-1-yl)-2-(2-methyl-4-oxo-(3H) quinazolin-3-yl) acetamide ( 8 ).
A mixture of the enaminone (7) (0.01 mol.) and 3-p-nitrophenyl-2-cyano acrylonitrile (0.01 mol.) in ethanol (20 ml) containing a few drops of trimethylamine (TMA) was heated under reflux for 5 hours. The formed solid product was filtered, dried and crystallized from absolute ethanol. Yield 63 %, m.p. 224-225 °C. IR (KBr) cm-1: 3443( NH ), 3317, 3189( NH2 ), 3014( CH Ar ) , 2948(CH Aliph.), 2184(C≡N), 1729, 1678, 1658 (3 C=O),1609, 1346 ( NO2 ) .1HNMR DMSO- d6 (δ ppm): 0.76 (s, 3H, CH3), 1.09 (s, 3H, CH3), 2.1 (s, 2H, CH2), 2.27 (s, 2H, CH2), 2.6 (s, 3H, CH3), 4.45 ( s, 1H, CH), 4.8 (s, 2H, NCH2), 6.6 (s, 2H, NH2 exchangeable), 7.5-8.11 (m, 8H, Ar-H), 11.2 (s, 1H, NHCO exchangeable). Ana. calcd for : (C29H27N7O5) C 62.93, H 4.88, N 17.72; found: C 62.92, H 4.90, N 17.71.
Synthesis of 3-[(5-oxo-1, 3, 4-oxadiazol-2-yl) methyl]-2-methyl-quinazolin-4(3H) one (9).
A mixture of the acid hydrazide (4) (0.01 mol.) and ethyl chloroformate (0.01 mol) in n-butanol (30 ml) was stirred under reflux for 20 hours. After cooling, the precipitate obtained was filtered off, washed with ethanol and crystallized from glacial acetic acid . Yield 58.5%, m.p = 360-363 °C. IR (KBr) cm-1: 3177 (NH), 3066(CH Ar), 2974, 2950 (CH Aliph.), 1711, 1696 (2C=O). 1HMNR DMSO- d6 (δ ppm): 2.54 (s, 3H, CH3), 4.8 (s, 2H, NCH2), 7.45 -8.1 (m, 4H, ArH), 10.6 (s, 1H, NH exchangeable). Ana. calcd for : (C12H10N4O3) C 55.81, H 3.90, N 21.70; found: C 55.25, H 4.12, N 22.10.
Synthesis of the potassium dithiocarbazate salt (10)
carbondisulphide(0.15 mole) was added to ice cold solution of potassium hydroxide (0.15 mole) in absolute ethanol containing (4) (0.1 mole). The mixture was diluted with absolute ethanol and stirred at room temp. for 12 hour. Dry ether 200 ml was added and separated solid was filtered and washed with ether. The product was employed in the next reaction without further purification. Yield nearly quantitative.
Synthesis of 2-[2-methyl-4-oxo-3(H)-quinazolin-3-yl]-N-(4-substituted phenyl-2-thiaoxothiazol-3-yl) acetamide (11 a-c).
To the compound (4) (0.01 mol.) in 1, 4-dioxane (30 ml), carbon disulphide (0.01 mol.) and potassium hydroxide (0.01mol.) in water (10 ml) were added. The reaction mixture was heated in boiling water bath for 1 hour then left to cool till 20 °C and to this cold solution of the mixture, the appropriate phenacyl bromide was added. The reaction mixture was stirred at room temperature over night. The solid product obtained after acidification with conc. HCL was collected by filtration and crystallized from absolute ethanol
(11a) Yield 85.5%; mp 216-218°C. IR (KBr, cm): 3170 ( NH ), 3061 ( CH Ar ), 2967, 2923 ( Aliph. ), 1681 (C=O), 1324 ( C=S).. 1HNMR(DMSO-d6, d ppm): 2.6 (s, 3H, CH3), 5.1 (s, 2H, CH2), 5.5 (s, 1H, =CH), 7.5-8.1 (m, 9H, ArH), 10.5(s, 1H, NH exchangeable ), Ana. calcd for : ( C20H16N4O2S2) C 58.80, H 3.95, N 13.72. ; found: C 58.42, H 4.3, N 13.51.
(11b) Yield 88.7%; mp 167-168°C. IR (KBr, cm): 3422 ( NH ), 3064 ( CH Ar), 2926 ( Aliph.), 1679 ( C=O ), 1393 ( C=S ).. 1HNMR(DMSO-d6, d ppm): 2.6 (s, 3H, CH3), 5.02 (s, 2H, CH2), 5.5 (s, 1H, =CH), 7.4-8.03 (m, 8H, ArH), 10.33 (s, 1H, NH exchangeable ), Ana. calcd for : (C20H15ClN4O2S2) C 54.23, H 3.41, N 12.65; found: C 58.42, H 4.3, N 12.61.
(11c) Yield 65%; mp 231-232°C. IR (KBr, cm): 3178 ( NH ), 3064 ( CH Ar), 2964 ( Aliph.), 1678 (C=O), 1340 ( C=S ). ), Ana. calcd for : (C20H15BrN4O2S2) C 49.58, H 3.09, N 11.57 ; found: C 49.25, H 3.15, N 11.4.
Synthesis of 3-[(5-thioxo-1, 3, 4-thiadiazol-3-yl) methyl] –2-methyl quinazolin-4(3H) one (13)
The potassium dithiocarbazate salt (10) (0.01 mol.) was added portion wise to ice cold conc. H2SO4 (7ml) while stirring, the reaction mixture was left overnight, then gradually added to crushed ice. The separated solid was filtered off and washed with water, then crystallized from absolute ethanol. Yield 40%, m.p. 256-7°C. IR (KBr) cm-1: 3001 ( CH Ar ), 2963, 2922 ( CH Aliph.), 2528 ( SH ), 1684( C=O ). 1HMNR DMSO-d6 (δ ppm): 2.54 ( s, 3H, CH3 ), 4.94 ( s, 2H, CH2 ), 7.52-8.12 ( m, 4H, ArH ), 10.6 ( s, 1H, NH exchangeable ). Ana. calcd for : (C12H10N4OS2 ) ,C 49.64,H 3.47, N 19.30 ; found: C 49.41, H 3.84, N 19.88.
Synthesis of 3-[(5-subustitutedthio-1, 3, 4-oxadiazol-2-yl) methyl] –2-methyl quinazolin-4(3H) one (15 a-f).and 3-[(5-subustitutedthio-1, 3, 4-thiadiazol-2-yl) methyl – 2-methyl quinazolin-4(3H) one(16 a-d).
A mixture of 12 (or 13) (0.01 mol.) and appropriate alkyl halide (0.01mol.) in DMF (20 ml) containing potassium carbonate (0.01 mol.) was stirred for 24 hours, water was added and the separated solid was filtered and crystallized from glacial acetic acid.
(15a): Yield 72.7%; mp 170-172°C. IR (KBr, cm-1): 3071, 3024 ( CH Ar ), 2996, 2965 ( CH Aliph.), 1674 ( C=O ). 1HNMR(CDCl3, δppm): 2.71 (s, 3H, CH3), 2.75 (s, 3H, CH3), 5.5 (s, 2H, CH2), 7.53-8.18 (m, 4H, ArH). Ana. calcd for : (C13H12N4O2S) C 54.16, H 4.16, N 19.40 ; found: C 54.58, H 4.2, N 19.31.
(15b): Yield 34%; mp 165-166°C. IR (KBr, cm-1): 3026 ( CH Ar ), 2997, 2923 ( Aliph.), 1674 ( C=O ). Ana. calcd for : (C14H14N4O2S) C 55.62, H 4.64, N 18.54; found: C 55.56, H 4.29, N 18.39.
(15c): Yield 36%; mp 128-129°C. IR (KBr, cm-1): 3029 ( CH Ar ), 2989, 2933 ( Aliph.), 1671 ( C=O ). 1HNMR(DMSO-d6 δppm): 2.64 (s, 3H, CH3), 4.47 (s, 2H, SCH2), 5.55 (s, 2H, NCH2), 7.23-8.13 (m, 8H, ArH). Ana. calcd for : (C19H16N4O2S) C 62.63, H 4.39, N 15.38 ; found: C 62.44, H 4.80, N 15.11.
(15d): Yield 50%; mp 304-305°C. IR (KBr, cm-1): 3212 ( NH ), 3063 ( CHAr ), 2992, 2925 ( Aliph.), 1733, 1674( C=O ).Ana. calcd for : (C20H17N5O3S) C 58.96, H 4.17, N 17.19; found: C 59.26, H 4.26, N 16.91.
(15e): Yield 31%; mp 238-239°C. IR (KBr, cm-1): 3222 ( NH ), 3063 ( CHAr ), 2988, 2826 ( Aliph.), 1735, 1687( C=O ). Ana. calcd for : (C21H19N5O4S) C 57.66, H 4.34, N 16.01 ; found: C 57.54, H 4.32, N 15.97.
(15f): Yield 45%; mp 275-277°C. IR (KBr, cm-1): 3166 ( NH ), 3067 ( CHAr ), 2994, 2939 ( Aliph.), 1728, 1673 ( C=O ).1HNMR(DMSO-d6 δppm): 2.39 (s, 3H, CH3), 3.83 (s, 3H, OCH3), 4.26 (s, 1H, SCH2), 4.85 (s, 2H, NCH2), 7.05-8.06 (m, 8H, ArH), 10.72 (s, 1H, CONH exchangeable ) Ana. calcd for : (C21H19N5O4S) C 57.66, H 4.34, N 16.01; found: C 58.03, H 4.87, N 15.67.
(16a): Yield 53%; mp 162-164°C. IR (KBr, cm-1): 3069 (CH Ar), 2993 (CH Aliph.), 1674(C=O). Ana. calcd for : (C13H12N4OS2) C 51.31, H 3.97, N 18.40; found: C 51.30, H 4.32, N 18.41.
(16b) Yield 61%; mp 190-191°C. IR (KBr, cm-1): 3263 (NH), 3075(CH Ar), 2980 ( CH Aliph.), 1667(2C=O). 1HNMR(DMSO-d6 δppm): 2.6 (s, 3H, CH3), 4.2 (s, 2H, sCH2), 5.6 (s, 2H, NCH2), 7.1-8.15 (m, 9H, ArH), 10.33 (s, 1H, CONH exchangeable ) Ana. calcd for : (C20H17N5O2S2) C 56.72, H 4.05, N 16.54; found: C 56.65, H 4.14, N 16.25.
(16c) Yield 58.2%; mp 211-212°C. IR (KBr, cm-1): 3223 (NH), 3064(CH Ar), 2988( CH Aliph.), 1734, 1675 (2C=O).Ana. calcd for : (C20H16ClN5O2S2) C 52.45, H 3.52, N 15.29; found: C 52.40, H 3.79, N 15.17.
(16d) Yield 61%; mp 207-208°C. IR (KBr, cm-1): 3335 (NH), 3055, 3013 (CH Ar), 2953,2920 (Aliph.), 1675 (2C=O). Ana. calcd for : (C21H19N5O3S2) C 55.62, H 4.22, N 15.44; found: C 55.63, H 4.22, N 15.24.
Synthesis of 3-[(4-Amino-5-thioxo-1, 2, 4-triazol-3-yl) methyl] – 2-methyl quinazolin-4(3H) one (14).
A mixture of potassium dithiocarbazate salt (10)(0.01 mol.) and hydrazine hydrate (98.99%, 0.02 mol.) in absolute ethanol (20 ml) was heated under refluxed for 8 hours, then cooled in room temperature and added to ice water. The mixture was neutralized with dil. HCl and the precipitate obtained was filtered off and crystallized from absolute ethanol to give compound (1) with yield 72% and m.p 150-152°C IR(KBr) cm-1: 3311-3284 ( NH2 ), 3187 ( NH ), 3064,3005 (CH Ar), 2979, 2928 ( CH Aliph.), 1690 ( C=O ).1HNMR DMSO-d6 (δ ppm): 2.60 (s, 3H, CH3), 5.39 (s, 2H, NCH2), 5.71 (s, 2H, NH2), 7.51-8.14 (m, 4H, ArH), 13.65 (s, 1H, NH exchangeable).Ana. calcd for : (C12H12N6OS) C 49.99, H 4.19, N 29.16; found: C 49.85, H 4.10, N 29.10.
Synthesis of 3-[(4-Arylideneamino-5-thioxo -1, 2, 4-triazol-3-yl) methyl] -2-methyl quinazolin-4(3H) one (17a,b).
A mixture of compound (14)(0.01 mol.) and the appropriate aromatic aldehyde (0.01 mol.) in ethanol / glacial acetic acid mixture (20 ml) was heated under reflux for 10-12 hours, most of the solvent was distilled off and the residue obtained was filtered and crystallized from glacial acetic acid
(17a): Yield 45%; mp 184-186°C. IR (KBr, cm-1): 3447 ( NH ), 3067 ( CH Ar ), 2934 ( CH Aliph.), 1666 ( C=O ). 1HNMR(DMSO-d6 δppm): 2.51 (s, 3H, CH3), 3.43, 3.88(2s, 6 H, 2CH3), 5.4 (s, 2H, NCH2), 7.12-8.18 (m, 8H, ArH), 8.8 (s, 1H, N=CH), 13.6 (s, 1H, NH exchangeable ) Ana. calcd for : (C21H21N7OS) C 60.14, H 5.01, N 23.38; found: C 60.22, H 5.46, N 23.38.
(17b): Yield 30%; mp 251-252°C. IR (KBr, cm-1): 3447 ( NH ), 3067 ( CH Ar ), 2934 ( CH Aliph.), 1666 ( C=O ). Ana. calcd for : (C19H15N6OSCl) C 55.33, H 3.64, N 20.38; found: C 55.33, H 3.63, N 20.51.
Synthesis of N-Phenyl-N’-[3-(2-methyl -4-oxo- quinazolin -3-yl) methyl- 5-thioxo -1, 3, 4-triazol-4-yl] thiourea (18 ).
To a solution of compound (14)(0.01 mol.) in DMF (20 ml), phenyl isothiocyanate (0.01 mol.) was added. The reaction mixture was stirred for 12 hours at room temperature, Water was added and the precipitate obtained was filtered and crystallized from absolute ethanol. Yield 83%, m.p. 184-6°C.IR (KBr) cm-1: 3222, 3172, 3122 ( 3NH ), 3004 (CH Ar), 2972 (CH Aliph.), 1678(C=O), 1324 ( C=S). 1HNMR DMSO-d6 (δ ppm): 2.57 ( s, 3H, CH3 ), 5.3 ( s, 2H, NCH2 ), 7.37-8.16 ( m, 8H, ArH ), 10.07 ( s, 1H, NH exchangeable), 10.4-10.6 ( broad, 2H, 2NH exchangeable ).Ana. calcd for : (C19H17N7OS2 ) ,C 53.88, H 4.05, N 23.15; found: C 53.68, H 3.82, N 23.46.
Synthesis of 3-[(6-Methyl- [1, 2, 4]-triazolo-[3, 4-b]-1, 3, 4-thiadiazol-3-yl) methyl]-2-methyl quinazolin-4(3H) one(19).
To a mixture of the compound (14)(0.01 mol) and acetic acid (0.52ml, 0.01 mol), phosphorus oxy chloride (10 ml) was added and the reaction content was refluxed for 7 hours on a water bath. The reaction mixture was slowly poured into crushed ice with stirring and neutralizing with solid sodium bicarbonate. The separated solid was filtered, washed with cold water, dried and crystallized from absolute ethanol. Yield 54%, m.p. 190-1 ° C. IR(KBr) cm-1 : 3056 ( CH Ar ), 2992, 2956 ( CH Aliph.), 1690 (C=O). 1HNMR CDCl3 (δ ppm): 2.27, 2.85 ( 2 s, 6H, 2CH3 ), 5.74 (s, 2H, NCH2), 7.43-8.24 ( m, 4H, ArH ).Ana. calcd for (C14H12N6OS) C 53.83, H 3.87, N 26.91; found: C 53.84, H 3.86, N 26.81.
Synthesis of 3-[(6-Mercapto-[1,2,4]-triazolo-[3,4-b]-1,3,4-thiadiazol-3-yl) methyl]-2-methyl quinazolin-4(3H) one (20).
The compound (14)(0.01mol) was dissolved in alcoholic solution of KOH (0.01 mol. in 20 ml absolute ethanol). Carbon disulfide (2ml) was added and the mixture was refluxed for 6 hours. Most of the solvent was evaporated and the residue was dissolved in 10% KOH solution and filtered. The cold filtrate was acidified with conc. HCl, the separated solid was filtered off, washed with water and crystallized from absolute ethanol. Yield 58.26%, m.p. 249-250 °C. IR(KBr) cm-1: 3429 ( NH ), 3001 ( CH Ar ), 2965, 2924 ( CH Aliph.), 1686 (C=O). 1HMNR DMSO- d6 (δ ppm): 2.6 (s, 3H, CH3), 5.4 (s, 2H, NCH2), 7.51-8.16 (m, 4H, ArH), 14.65 (br, 1H, NH exchangeable).Ana. calcd for : (C13H10N6OS2) C 47.26, H 3.05, N 25.44; found: C 47.55, H 3.29, N 25.80.
Synthesis of 3-[(6-P-Chlorophenyl-7H-[1, 2, 4]-triazolo-[3, 4-b]-1, 3, 4-thiadiazin-3-yl) methyl]-2-methyl quinazolin-4(3H) one(21).
A mixture of (14)(0.01 mol.) and P-chlorophenacyl bromide (0.01mol.) in absolute ethanol (20 ml) was stirred under reflux for 8 hours. After cooling, the reaction mixture was neutralized with ammonium hydroxide solution and the liberated free base was filtered, washed with water and crystallized from absolute ethanol. Yield 43%, m.p. 207-9°C.IR (KBr) cm-1: 3040 ( CH Ar ), 2920, 2808 ( CH Aliph.), 1701( C=O ). 1HNMR DMSO-d6 (δ ppm): 2.6 ( s, 1H, CH3 ), 3.4 ( s, 2H, SCH2 ), 5.5 ( s, NCH2 ), 7.6-8.2 ( m, 8H, ArH ). Ana. calcd for :(C20H15ClN6OS) C 56.80, H 3.58, N 19.87; found: C 56.68, H 3.55, N 19.84.
Anti-inflammatory activity
Mature albino rats of both sex weighing 150-200 gm were used. They were divided into 10 equal groups (each of 5). The first group was left as a control while the second one was injected (I.P.) with celecoxib at dose of 0.9 mg/100 gm. The tested compounds were injected (I.P.) to 8 groups at the same dose level. One hour later, oedema in the right hind paw was induced by injection of 0.1 ml of 10% carrageenin. The thickness of the paw was measured 1,2,3 and 4 hours after carrageenin injection to determine the Anti-inflammatory activity of the tested compounds.
Gastric ulcerogenic effect
Male albino rats weighing 150-200 gm were fasted for 12 hours prior to drug adminstration. Water was supplied ad-libitum. The animals were divided into 10 equal groups (each of 5). The first group received 1% gum acacia (suspending vehicle) orally once a day and left as a control, whereas, the second group received indomethacin at a dose of 0.9 mg/ 100 gm/ day orally using metallic stomach tube. Groups from 3rd to 10th received the tested compounds at the same dose. The drugs were administered once a day for 3 successive days. The animals were killed by overdose of ether 6 hours after the last dose. The stomachs were removed, opened along the greater curvature and examined for ulceration. The number and severity of discrete areas of damage in the glandular mucosa were scored. The ulcer score was calculated according to the 1 to 5 scoring system of Wilhelemi and Menasse- Gdynia (1972).
Stomach ulceration was expressed in terms of ulcer index ( U.I = mean ulcer score of a groups of animals similarly treated X % of ulcerated animals of this group(Pauls et al., 1947).
Molecular docking
All the molecular modeling studies were carried out on an Intel Pentium 1.6 GHz processor, 512 MB memory with Windows XP operating system using Molecular Operating Environment (MOE 2005.06; Chemical Computing Group,Canada) [17] as the computational software. All the minimizations were performed with MOE until a RMSD gradient of 0.05 Kcal mol-1 Å-1 with MMFF94X force-field and the partial charges were automatically calculated. Docking studies of the inhibitors were performed by MOE (Molecular Operating Environment) using murine cyclooxygenase-2 co-crystallized with selective inhibitor, celecoxib (Protein Data Bank code PDB ID: 6COX) as a template. The enzyme was prepared for docking studies where: (i) ligand molecule was removed from the enzyme active site. (ii) Hydrogen atoms were added to the structure with their standard geometry. (iii) MOE Alpha Site Finder was used for the active sites search in the enzyme structure and dummy atoms were created from the obtained alpha spheres. (iv) The obtained model was then used in predicting the ligande-enzyme interactions at the active site.
References
- J. Vane, Towards a better aspirin, Nature 367, (1994)215–216.
- J.J. Talley, Selective inhibitors of cyclooxyganase-2, Exp. Opin. Ther. Patents(1997)755–62.
- L.A. Sorbera, P.A. Leeson, J. Castafier, Rofecoxib, Drugs of the future 23 (1998)1287–1296.
- B.Audeval,P. Bouchacourt, J. Rondier. Comparative study of diproqualone-ethenzamide versus glafenine for the treatment of rheumatic pain of gonarthrosis and coxarthrosis. Gazette medicale de France; 95(25): (1988)7072.
- SV. Bhandari, KG. Bothara, MK. Raut, AA. Patil and AP. Sarkate. Bioorganic and Medicinal Chemistry.; 16(4) : (2008)1822-31.
- FA. Omar, NM. Mahfouz, MA. Rahman. Eur. J. Med. Chem., 31(10), (1996)819-825 .
- L.S. Varandas , C.A.M. Fraga , A.L.P. Miranda and E.J. Barreiro. Letters in Drug Design & Discovery, 2, (2005)62-6.
- K. Andanappa, B. Mahesh, K.Palkar, S. Thippeswamy and J. Boreddy. Bioorganic & Medicinal Chemistry, 16, (1), 276-283(2008).
- B. Tozkoparan, E. Küpeli, E. Yeşilada and M. Ertan. Bioorganic & MedicinalChemistry, 15(4),(2007)1808-1814.
- U. Salgın-Gökşen, N. Gökhan-Kelekçi, Ö. Göktaş, Y. Köysal, E. Kılıç, Ş. Işık, G. Aktay, M. Özalp. Bioorganic & Medicinal Chemistry, 15(17), (2007)5738-5751.
- M. T. Bogert and H. A. Seil. J. Am. Chem. Soc.29, (1905)217.
- J. Ponda, S. Srinivas. J. Indian chem. Soc., 79(9), (2002)770-771;.
- Ana Berechet. Farmacia (Bucharest) 15(5), (1967)287-94, (Rom.).
- A. R. Desai and K. R. Desai. J. Heterocyclic Chem., (2005)42,955-9.
- C. Winter, E. Rilsley and G.W. Nuss; Pro. Soc. Exp. Biol. Med., 3, (1962)544.
- H. Ikuta, H. Shirota, S. Kobayashi, Y. Yamagishi, K. Yamada, I. Yamatsu, K. Katayama, J. Med. Chem. 30, (1987)1995-1998.
- Molecular Operating Environment (MOE), Version 2005.06, Chemical Computing Group, Inc., Montreal, Quebec, Canada, 2005.<http://www. chemcomp.com.
Accepted on: January 01, 2009








![]()
![]()
