

How to Cite | Publication History | PlumX Article Matrix
Individual Domain Stability of Klenow-Like DNA Pol I ITB-1 Based on Molecular Dynamic Simulation
Hira Helwati1,2, Santi Nurbaiti2, Rukman Hertadi2, Fida Madayanti Warganegara2 and Akhmaloka2*
1Department of Chemistry, University of Syahkuala, Banda Aceh Indonesia.
2Biochemistry Research Group, Faculty of Mathematics and Natural Sciences, Institute, Teknologi Bandung, Jln Ganesha 10 Bandung - 40132 Indonesia.
Corresponding Author E-mail:loka@chem.itb.ac.id
ABSTRACT: The structural stability of individual domains in Klenow-like DNA polymerase I ITB-1 were carried out by molecular dynamics simulation. Based on Ca-root-mean square deviation (RMSD) at 300K, supported by root-mean-square fluctuation (RMSF), secondary structure and solvent accessible surface area (SASA) analysis suggested that the Klenow-like DNA Pol I ITB-1, 3'®5' exonuclease and 5’®3’ polymerase domains did not show significant change in the proteins structure during 10 ns MD simulation. These results suggested that both domains were stable and may have an independent folding at 300K. However, RMSD analysis at higher temperature (360 K) suggested that polymerase domain was the most unstable, indicated by increasing the RMSD value of the domain was faster than that the other two protein models. RMSD analysis at 360 K with extended times of simulation suggested that the structure of 3'®5' exonuclease domain required longer time to unfold than that polymerase domain. The MD simulation at 360 K suggested that interaction between 3’®5’ exonuclease and 5’®3’ polymerase domains have significant role in maintaining thermal stability of the whole structure of Klenow-like DNA Pol I ITB-1.
KEYWORDS: Klenow-like DNA Pol I ITB-1; protein stability; molecular dynamic simulation; imdividual domain
Download this article as:| Copy the following to cite this article: Helwati H, Nurbaiti S, Hertadi R, Warganegara F. M, Akhmaloka.Individual domain stability of klenow-like DNA Pol I ITB-1 based on molecular dynamic Simulation. Biosci Biotechnol Res Asia 2010;7(2) |
| Copy the following to cite this URL: Helwati H, Nurbaiti S, Hertadi R, Warganegara F. M, Akhmaloka.Individual domain stability of klenow-like DNA Pol I ITB-1 based on molecular dynamic Simulation. Biosci Biotechnol Res Asia 2010;7(2). Available from: https://www.biotech-asia.org/?p=8796 |
Introduction
DNA polymerases are present in all organisms and involved in genomic replication and repair. Three types of DNA polymerases have been isolated from Escherichia coli. DNA polymerase (Pol) I is the most abundant DNA Pol found in E. coli and was also the first polymerase characterized1. It has three distinct structural domains: the N-terminal 5’à3’ exonuclease, the central 3’à5’ exonuclease and the C-terminal 5’à3’ polymerization domains2. The 5’à3’ exonuclease degrades duplex DNA from its 5’ end. It enables DNA polymerase I to remove RNA primers of Okazaki fragments and fill with DNA sequence. The 5’à3’ exonuclease is also capable of excising DNA lesions, such as thymine dimers, during DNA repair. The 3’à5’ exonuclease degrades single or double stranded DNA from their 3’ ends. This ability to remove mismatched nucleotides provides DNA replication a proofreading activity2. Three sequence motifs of 3’à5’ exonuclease sub-domains (Exonuclease I, II and III) have been identified by sequence comparisons3,4 and mutagenesis experiments5. A number of prokaryotic and eukaryotic DNA polymerases have an intrinsic 3’à5’ exonuclease activity6. Structural studies5,7,8 indicate that the Exo catalytic site is located in an N-terminal domain that is separated from the polymerase domain. The structure of the Exo active site is highly conserved. Two divalent metal ions are present, and they are coordinated by the amino acids of three signature Exo motifs conserved throughout all families of DNA Pol3.
A few moderately thermostable DNA polimerase have been isolated and purified from thermophilic Bacillus species9, such as Bst DNA polymerase was isolated from B. stearothermophilus10-12 and Bca DNA polymerase from B. caldotenax13. Coding sequence (CDS) of local DNA pol I gene from West Java, namely DNA pol I ITB-1, was isolated from Geobacillus thermoleovorans. The gene has been cloned and expressed in E. coli, consists of 2682 nucleotides that encodes 876 amino acid residues14. Crude extracts of the expressed protein showed optimum activity at 65oC (338 K) and pH 7.4. Early thermal stability study of Klenow-like DNA Pol I ITB-1 by MD simulation at higher temperature (400 and 500 K) revealed that the thermostability region was distributed in the whole protein structure15. Furthermore, MD simulation at 350 K suggested that the early stage of unfolding process of the protein was occurred by disruption of the interface region between 3’à5’ exonuklease and 5’à3’ polymerase domains16. Moreover, the domain motion of Klenow-like fragment in absent of substrat also analyzed by (MD) simulation sugested that 3’à5’ exonuclease and 5’à3’ polymerase domains exhibited anticolerated motion17. However the role of inter domain interaction for thermal stability of individual domain have not been known yet.
The special folding problem of the multidomain proteins is whether domains or even subdomains can refold independently and associate only at a late stage of folding or whether they associate at an early stage and refold only in the presence of each other in a cooperative manner18. Regarding the stability and folding of domains in modular proteins, in vitro studies have shown that there are distinct and individual differences with respect to the interactions between domains and their mutual stabilizing or destabilizing effects. Comparing at this point the individual contributions of domains to the overall stability of modular proteins, commonly mutual stabilization has been observed. In certain cases, folding experiments gave clear evidence that such stabilization may provide `assistance’ in the self-organization of proteins, once more stressing their multifunctional nature: domains not only diversify functionality and promote evolution, but at the same time they allow the folding efficiency and stability of proteins to be optimized19.
Large theoretical and experimental researches are devoted to understand how protein folds into its native structure20-22. Molecular dynamics (MD) simulation is a powerful tool to complement the experimental results with atomic and instantaneous details24-23. Nevertheless, direct atomistic simulations of protein folding are currently unfeasible since the biological activity (ms-s) is far beyond the reach of such simulations (~ns). On the other hand, computations have shown that unfolding may capture the essential aspects of the folding process25-26 and high temperature can accelerate the unfolding process without affecting the pathway27. Since there were very limited information concerning thermal stability of Klenow-like DNA Pol I structure, particularly on the stability of individual structure of the domains and their role in whole protein stability, here we reported the MD simulation to study stability of the individual domains of Klenow-like DNA Pol I ITB-1.
Material and Methods
Construction of Structural Model
The initial three-dimensional structure of the Klenow-like DNA Pol I ITB-1, 3’à5’ exonuclease and 5’à3’ polymerase domains were constructed based on homological protein structural modeling from SWISS-MODEL program28 (Arnold et al., 2006) where the highest homology template was found by using interproScan program29 (Zdobnov et al., 2001).
Simulation Methods
Amber version 9.0 program30 was used for the molecular dynamic simulation. Parm99 + frcmod ff03 was used for the simulation. The implicit solvent (igb = 5) was used to describe the solvation effects in MD simulation. The mbondi2 radii were set, and the reaction field cut off = 15 was employed to speed-up the calculation of effective Born radii. The temperature and pressure were kept constant during the simulation. The trajectories and coordinate of the protein were saved every 2 fs for structural analysis. Simulation image of protein was generated by visual molecular dynamic (VMD) software31.
Analysis of MD Trajectories
Analysis of the MD coordinate trajectories of the simulations were performed by calculating several structural parameter over time, such as C-a root-mean-square-deviation (RMSD), root-mean-square-fluctuation (RMSF), solvent accessible surface area (SASA) and percentage of secondary structure (% SS) that were calculated within software package VMD.
Results
Structure of Klenow-like DNA Pol I ITB-1 and Each Individual Domain
Based on template homology using interproScan program, the sequence of well-characterized crystal structure of Bacillus fragment (BF) from Bacillus stearothermophilus DNA polymerase I (PDB entry 1nK4 chain A) was found as the highest sequence identity (98%) to the fragment of DNA Pol I ITB-1 from amino acid residue at 297 to 876, containing 3’à5’ exonuclease (residue 297 to 468) and 5’à3’ polymerase regions (residue 469 to 876). The resulting three dimensional structure of the Klenow-like DNA pol I ITB-1 and the inividual 3’à5’ exonuclease and 5’à3’ polymerase domains using Swiss-model modeling program based on 1nK4A PDB template, showed 99% identity for all protein models. The 3D structure of Klenow-like DNA Pol I ITB-1 and both individual domains generated from the comparative modeling almost resemble with the structure of its template with RMSD value at 1.46 Å (Fig1). All structural feature in the polymerase domain, such as palm, thumb and fingers were similar to those found in known DNA Pol I structures, indicated that the structural model of DNA Pol I ITB-1 was successfully determined and reliable to used as an initial coordinate for molecular dynamic simulation in this study.
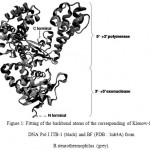 |
Figure 1: Fitting of the backbond atoms of the corresponding of Klenow-like DNA Pol I ITB-1 (black) and BF (PDB : 1nk4A) from B.stearothermophilus (grey).
|
Stability of Proteins at 300 K
The MD simulations were carried out at temperature 300 K in implicit solvent, using generalized Born solvation model. MD simulation at this temperature was intended to assess the conformational stability of DNA Pol I ITB-1 at room temperature. The RMSD that represented the structural change during the simulation32 showed that the structures of the enzyme and its individual domains were stable during 10 ns simulation at 300K. The RMSD of the whole structure and polymerase domain were found at around 3-5 Ǻ from the initial structure while 3’à5’ exonuclease domain was found at around 3 Ǻ (Fig 2). Alignment between the structures after 10 ns and 0 ns, showed that the average RMSD difference were about 4.85 Å for Klenow-like DNA Pol I ITB-1, 5.75Å for 5’à3’ polymerase and 3.17Å for 3’à5’ exonuclease domains (Fig 3). Further analysis of the SASA value showed that there were no significant changes on the tertiary structure during the simulation for all of the proteins models (Fig 4). The secondary structure percentage (%SS) of Klenow-like DNA Pol I ITB-1 and 5’à3’ polymerase domain showed stable until the end of simulation (10 ns), however the helix composition of 3’à5’ exonuclease domain was decreased at the end of simulation (Fig 5). The RMSF value represents the flexibility profile of individual amino acid residues in the protein models, showed that there were some extreme values exhibited by amino acid residues at certain position as indicated by the appearance of some peaks in RMSF curve (Fig 6). The peaks position were located in both domains, where residues at 412 to 418 and 430 to 434 were more unstable region in 3’à5’ exonuclease domain, while the 5’à3’ polymerase domain have unstable residues at 530 to 566 located at thumb sub-domain and residues at 778 to 781 on fingers sub-domain. The fluctuation of amino acid residues in both domains almost similar to that the Klenow-like DNA Pol I ITB-1 residues, indicated that individual domain were stable during the simulation. The stability of the protein structures at room temperature justified the reliability of the implicit solvent system used in this study16.
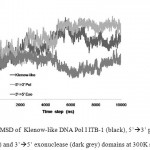 |
Figure 2: RMSD of Klenow-like DNA Pol I ITB-1 (black), 5’à3’ polymerase (light grey) and 3’à5’ exonuclease (dark grey) domains at 300K simulation.
|
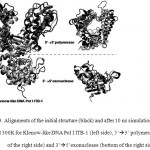 |
Figure 3: Alignments of the initial structure (black) and after 10 ns simulation (grey) at 300K for Klenow-like DNA Pol I ITB-1 (left side), 5’à3’ polymerase (top of the right side) and 3’à5’exonuclease (bottom of the right side)
|
 |
Figure 4: Total and apolar SASA values at 300 K for Klenow-like DNA Pol I ITB-1, 5’à3’ polymerase domain and 3’à5’ exonuclease domain.
|
 |
Figure 5: Percentage of secondary structures (α-helix and β-sheet) at 300 K for Klenow-like DNA Pol I ITB-1, 5’à3’ polymerase domain and 3’à5’ exonuclease domain.
|
 |
Figure 6: The RMSF value as function of residue number at 300K for Klenow-like DNA Pol I ITB-1 (residue 297 to 876, black), 3’à5’exonuclease domain (residue 297 to 468, green) and 5’à3’ polymerase domain (residue 469 to 876, red). |
Simulation of Model Proteins at 360 K
The stability of each domains, 3’à5’ exonuclease and 5’à3’polymerase, were decrease when the protein models were simulated at higher temperature and longer times. The RMSD analysis of the domains at 360 K during 22 ns of simulation, showed that the 5’à3’ polymerase domain unfolded faster than two other protein models, indicated by sharp increase of the RMSD value at 8 to 11 ns while RMSD of 3’à5’ exonuclease domain increase sharply after 14 ns. On the other hand, the Klenow-like DNA Pol I ITB-1 remains stable during 4 to 20 ns of simulation (Fig 7). The RMSD values of the proteins were visualized by structure alignments of the initial structures against the structures after 22 ns of simulation, the result showed that the structures of each domain were unstable compare to the Klenow-like structure. The 5’à3’polymerase and 3’à5’ exonuclease domains exhibited RMSD value at 17.9 Å and 18.6 Å, respectively, while the Klenow-like DNA Pol I ITB-1 was 13.9 Å (Fig 8). Further analysis of the flexibility of the individual amino acid residues in the proteins at 360 K by the RMSF analysis showed that the individual domains were more fluctuate than that the Klenow-like structure, indicated by higher RMSF value of the individual domain residues compare to the Klenow-like residues (Fig 9). The RMSF values also indicated that the fluctuative region for both separated domain were increased, where the residues 530 to 566 (thumb sub-domain) were the most unstable region. The RMSF value was converting into β-factor to clarify the location of flexible location in the proteins and the result was mapped into the structure of protein as shown in Fig 10. It shown that the unstable regions in the each domains structure were increased compared to the whole structure of Klenow-like fragment. The MD simulation at 360 K suggested that the individual domain, 5’à3’polymerase and 3’à5’ exonuclease domains separately were unstable at high temperature.
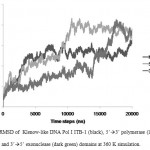 |
Figure 7: RMSD of Klenow-like DNA Pol I ITB-1 (black), 5’à3’ polymerase (light grey) and 3’à5’ exonuclease (dark green) domains at 360 K simulation.
|
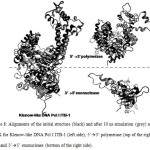 |
Figure 8: Alignments of the initial structure (black) and after 10 ns simulation (grey) at 360 K for Klenow-like DNA Pol I ITB-1 (left side), 5’à3’ polymerase (top of the right side) and 3’à5’ exonuclease (bottom of the right side). |
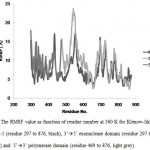 |
Figure 9. The RMSF value as function of residue number at 360 K for Klenow-like DNA Pol I ITB-1 (residue 297 to 876, black), 3’à5’ exonuclease domain (residue 297 to 468, dark grey) and 5’à3’ polymerase domain (residue 469 to 876, light grey). |
 |
Figure 10: Thermostablity maps of the Klenow-like DNA Pol I ITB-1 (A), 5’à3’ polymerase (B) and 3’à5’ exonuclease (C) based on β-factor value of the 360 K simulation.
|
Discussion
The understanding of the folding mechanisms of single domain proteins is an essential step in the understanding of protein folding in general. Theoretical studies lead to better understanding of experimental results providing easy-to-interpret structural models. Domains of DNA Pol I have distinct roles in DNA replication indicated by each domain showed independent stability and folding. Crystal structure analysis of Klenow fragment showed that the enzyme consist of two domains33. Analysis of this structure suggests that the active site of the 3’à5′ exonuclease and 5’à3 ‘ polymerase lied separately in each domain5. Moreover Taq Pol I which lost of the N-terminal domain (approximately 289 amino acid residues), showed an increase on thermostability compared to that the whole enzyme34. The result suggests that 5’à3’ exonuclease and Klenow-like fragment are two independent structures, and thermostability of Klenow-like fragment may not depend on 5’à3’ exonuclease domain.
In this research, we investigated the effect of high temperature on the structure of Klenow-like DNA Pol I ITB-1 dan its individual domains. By comparing the result at 300 K and 360 K, showed that high temperature condition was significantly affect the stability of 5’à3’polymerase and 3’à5’ exonuclease domains (Fig 2 and 7). However, at 300 K, the structures of both domains were stable, as same as Klenow-like DNA Pol I ITB-1 (Fig 2). Based on the RMSF analysis of the individual domains at 300K showed that the amino acid residue in both domain have similar fluctuation with the Klenow-like residues (Fig 6.) suggested that both domains have independent folding at 300 K.
In contrast at 360 K, both domains, 5’à3’polymerase and 3’à5’ exonuclease domains, were lost their stability. The RMSD of both protein models up to 22 ns of simulation at 360 K showed that the structure of individual domains were significantly difference compared to that the Klenow-like structure (Fig 7). The instability of each domain was also shown by higher fluctuation of amino acid residues compared to that the Klenow-like fragment. The RMSD profile during the simulation at 360 K showed that the 5’à3’ polymerase unfolded faster than that the 3’à5’ exonuclease domain (Fig 7). This phenomena are supported by a few experimental data. There were some difficulties in purifying the expressed of the C-terminal protein (5’à3’ polymerase domain) of the Klenow fragment 35-37, and the activity of 5’à3’ polymerase domain without the present of 3’à5’ exonuclease domain was decreased 10 fold compared to that the Klenow fragment37. These results suggested that the 5’à3’ polymerase domain need the present of 3’à5’ exonuclease domain to maintain its stability.
The result at 360 K suggested that the interaction between 5’à3’polymerase and 3’à5’ exonuclease have an important role in maintaining the stability of the Klenow-like DNA Pol I ITB-1 fragment. The interactions of both domains were needed to stabilize the structure of the Klenow-like fragment. However, each domain was unstable in each individual structure. All the results above were in agreement with our previous results16 that provided information about the role of interface domain interaction on thermal stability of Klenow-like DNA Pol I ITB-1. The result showed that early unfolding of the Klenow-like DNA Pol I ITB-1 was initiated at the interface region between 5’à3’ polymerase and 3’à5’ exonuclease domains. It has been shown that there are two salt bridges (Lys374-Glu489 and Lys381-Glu487) between 5’à3’polymerase and 3’à5’ exonuclease domains that taking a role in tightening the contact of the two domains16.
Conclusion
The structure of Klenow-like DNA Pol I ITB-1 is more stable compared to that the individual 3’à5’ exonuclease and 5’à3’ polymerase domains based on the MD simulations. Interaction between 3’à5’ exonuclease and 5’à3’ polymerase domains stabilized the fusion structure of Klenow-like DNA Pol I.
Acknowledgment
This research was partially support by Competitive Grant (Hibah Bersaing) from Ministry of National Education, Republic of Indonesia to FMW, and Research Grant from Institut Teknologi Bandung to RH and BPPS scholarship program to HH.
References
- Konberg A., Lieberman I., and Simms E.S., Enzymatic synthesis and properties of 5’-phosphoryl prophosphatase, Biochem. Chem., 215(1): 389-402 (1955).
- Kornberg A., and Baker T.A., DNA Replication, 2nd W.H. Freeman and Company. New York. (1992).
- Blanco L., Bernad A., Blasco M.A., and Salas M., A General Structure for DNA-dependent DNA Polymerase, Gene, 100:27-38 (1991).
- Morrison A., Bell J.B., Kunkel T.A, and Sugino, A., Eukaryotic DNA polymerase amino acid sequence required for 3′à5′ exonuclease activity. Natl. Acad. Sci. U.S.A., 88:9473–9477 (1991).
- Derbyshire V., Pinsonneault J.K., and Joyce C.M., Structure-function of 3’à5′ exonuclease of DNA polymerase, Methods in Enzymology, 32:363-384 (1995).
- Jin Y.H., Obert R., Burgers P.M.J., Kunkel T.A., Resnick M.A., and Gordenin D.A., The 3’à5’ exonuclease of DNA polymerase δ can substitute for the 5’ flap endonuclease Rad27 yFen1 in processing Okazaki fragments and preventing genome instability. Natl. Acad. Sci. U.S.A., 98(9):5122-5127 (2001).
- Steitz T.A., DNA Polymerase: Structural Diversity and Common Mechanisms, Biol. Chem., 274:17395-17398 (1999).
- Shamoo Y., Steitz T.A., Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex, Cell, 99: 155-166 (1999).
- Perler F.B., Kumar S., and Kong H., Thermostable DNA Polymerases, Protein Chem., 48: 377-435 (1996).
- Stenesh J., Roe R.A., DNA polymerase from mesophilic and thermophilic bacteria. Purification and properties of DNA polymerase from Bacillus licheniformis and Bacillus stearothermophilus. Biophys. Acta. 272:156-66 (1972).
- Kaboev O.K., Luchkina L.K., Akhmedov A.T., Bekker M.L., Purification and properties of deoxynucleic acid polymerase fro Bacillus stearothermophilus. Bacteriol. 145(1):21-6 (1981).
- Sellman E., Schroder K.L., Knoblich I.M., Westermann P., Purification and characterization of DNA polymerase from Bacillus J. Bacteriol. 174: 4350-5 (1992).
- Uemori T., Ishino Y., Fujita K., Asada K., Kato I., Cloning of the DNA polymerase gene of Bacillus caldotenax and characterization of the gene product. Biochem. 113: 401-10 (1993).
- Akhmaloka, Pramono H., Ambarsari L., Susanti E., Nurbaiti S., and Madayanti F., Cloning, Homological Analysis, and Expresion of DNA Pol I from Geobacillus thermolevorans. J. Integ. Biol., 1(3):206-215 (2007).
- Hertadi R., Nurbaiti S., Akhmaloka., Molecular dynamics analysis of thermostable DNA Pol I ITB-1. Indonesia, 1(3): 101-4 (2007).
- Nurbaiti S., Hertadi R., Martoprawiro M.A., and Akhmaloka., The Role of Interface Domain Interactions on Thermal Stability of DNA polymerase I ITB-1, The Open Struc. Biol. J., 3: 16-25 (2009).
- Nurbaiti S., Nagao H., Saito H., Hertadi R., Martoprawiro M.A., and Akhmaloka., Domain Motions of Klenow-like DNA polymerase I ITB-1 in the Absence of Substrate, J. Integ. Biol., 9(2):104-110 (2010).
- Szilágyi A., and Závodszky P., Structural differences between mesophilic, moderately thermophilic dan extremely thermophilic protein subunits: Results of a comprehensive survey, Structure, 8(5): 493-504 (2000).
- Jaenicke R., Stability and folding of domain proteins. Biophysics & Mol. Biol., 71:155-241 (1999).
- Onuchic J.N., Luthey-Schulten Z., and Wolynes P.G., Theory of protein folding: the energy landscape perspective. Rev. Phys. Chem., 48:545-600 (1997).
- Dobson C.M., and Karplus M., The fundamental of folding: bringing together theory and experiment, Opin. Struct. Biol., 9: 92-101 (1999).
- Alm E., Baker D., Matching theory and experiment in protein folding. Opin. Struct. Biol., 9:189-196 (1999).
- Duan Y., Kollman P.A., Pathways to a Protein Folding Intermediate Observed in a 1-Microsecond Simulation in Aqueous Solution. Science, 282(5389):740-744 (1998).
- Garcia A.E., Onuchic N., Folding a Protein in a Computer: An Atomic Description of the Folding/Unfolding of Protein A. Proc. Natl. Acad. Sci. U.S.A., 100(24): 13898-13903 (2003).
- Brooks C.L., Simulations of Protein Folding and Unfolding. Opin. Struct. Biol., 8(2):222-226 (1998).
- Fersht A.R., and Daggett V., Protein Folding and Unfolding at Atomic Resolution. Cell, 108(4): 573-582 (2002).
- Day R., Bennion B.J., Ham , and Daggett V., Increasing Temperature Accelerates Protein Unfolding Without Changing the Pathway of Unfolding. J. Mol. Biol., 322(1):189-203 (2002).
- Arnold K., Bordoli L., Kopp J., and Schwede T., The SWISS-MODEL Workspace: A web-based environment for protein structure homology modeling, Bioinformatics, 22:195-201 (2006).
- Zdobnov E.M., and Apweiler R., InterProScan – an integration platform for the signature-recognition methods in InterPro. Bioinformatics, 17: 847-848 (2001).
- Case D.A., Darden T.A., Cheatham T.E., Simmerling C.L., Wang J., Duke R.E., Luo R., Merz K.M., Pearlma D.A., Crowley M., Walker R.C., Wang W., Wang B., Hayik S., Roitberg A., Seabra G., Wong K.F., Paesani F., Wu X., Brozel S., Tsui V., Gohlke H., Yang L., Tan C., Mongan J., Homak V., Cui G., Beroza P., Mathews D.H., Schafmeister C., Ross W.S., and Kollman, P.A. AMBER 9, University of California, San Fancisco, (2006).
- Humphrey W., Dalke A., and Schulten K., VMD: visual molecular dynamics, Mol. Graph., 14(1), 33-38, 27-28 (1996).
- Becker O.M., MacKerrel A.D., Roux B., and Watanabe M., Computational Biochemistry dan Biophysics, Marcel Dekker, Inc., New York, (2001).
- Ollis D.L., Brick P., Hamlin K.M., Xuong N.G., and Steitz T.A., Structure of large fragment of Escherichia coli DNA polymerase I complexed with dTMP, Nature, 313:762-766 (1985).
- Lawyer F.C., Stoffel S., Saiki R.K., Mymbo K., Drummond R., and Gelfand D.H., Isolation, Characterization and Expresstion in coli of the DNA Polymerase Gene from Thermus aquaticus, J. Biol. Chem, 264:6427- 6437 (1989).
- Gribskov M., and Burgess R.R., Overexpression and purification of the sigma subunit of Escherichia coli RNS polymerase, Gene, 26:109-118 (1983).
- Scheuermann R.H., and Echols H., A separate editing exonuclease for DNA replication: the e subunit of Escherichia coli DNA polymerase III holoenzyme. PNAS, 81:7747-7751 (1984).
- Freemont P.S., Ollis D.L., Steitz T.A., and Joyce C.M., A domain of Klenow fragment of Escherichia coli DNA Polymerase I has polymerase but no exonuclease activity, Protein Struct., Funct. Genet., 1:66-73 (1986).

This work is licensed under a Creative Commons Attribution 4.0 International License.



