

How to Cite | Publication History | PlumX Article Matrix
Rajendra Jangde1*, Sanjay Daharwal1, Deependra Singh1 and Ram Kumar Sahu2
¹University Institute of Pharmacy, Pt. Ravishankar Shukla University, Raipur - 492 010 India.
²Oriental College of Pharmacy, Raisen Road, Bhopal - 462 021 India.
Corresponding Author E-mail: rjangdepy@gmail.com
ABSTRACT: Prolonged-release microcapsules of Ampicillin Hydrochloride (AH) were prepared by employing ethyl cellulose as a polymer in various ratios of 1:1, 1:3 & 1:4, by emulsion solvent diffusion technique. Scanning electron microscope photographs of samples revealed that all prepared microcapsules were almost spherical in shape and have a slightly smooth surface and evaluated for particle size, shape, flow properties, wall thickness, drug encapsulation efficiency and in vitro release performance. The drug carrier interactions were investigated in solid state by FT-IR spectroscopy. The present study concerned with the development and characterization of ampicillin microcapsules prepared by thermal change method using different ratios (1:1, 1:3 and 1:4) of ethyl cellulose in order to select the best microcapsule formulation with a good encapsulation efficiency and drug release profile. The obtained microcapsules were discrete, spherical with free flowing properties. The encapsulation efficiency was found to be in the range of 66.17-72.99%. The in vitro release profile of Ampicillin indicates that all the batches of microcapsules showed controlled and prolonged drug release over an extended period of 12 h. Drug release from the microcapsules (F1 and F2) was 56-51 % in first 6 hours, with the initial burst of nearly 50% within one hour. Drug release from microcapsules (F1 and F2) was 60-56% and sustained up to 8 h with initial burst of 50-54 % in first 6 h, resulted with increase in cross-linking time for 5-6 hours, Drug release from microcapsules (F2 and F3) sustained the drug release up-to 12 hours, The therapeutic effect of drugs that have a short biological half-life may be enhanced by formulating them as extended- or sustained-release dosage forms. Extended- and sustained-release dosage forms prolong the time that systemic drug levels are within the therapeutic range and, thus, reduce the number of doses the patient must take to maintain a therapeutic effect, thereby increasing compliance.
KEYWORDS: Microcapsules; Ampicillin; Ethyl cellulose; Sustain release
Download this article as:| Copy the following to cite this article: Jangde R, Daharwal S, Singh D, Sahu R. K. Development and Characterization of Ethyl Cellulose Coated Microcapsules for Controlled Release of Ampicillin. Biosci Biotech Res Asia 2011;8(2) |
| Copy the following to cite this URL: Jangde R, Daharwal S, Singh D, Sahu R. K. Development and Characterization of Ethyl Cellulose Coated Microcapsules for Controlled Release of Ampicillin. Biosci Biotech Res Asia 2011;8(2). Available from: https://www.biotech-asia.org/?p=9565 |
Introduction
The science of drug delivery may be described as the application of chemical and biological principles to control the in vivo temporal and spatial location of drug molecules for clinical benefit1. When drugs are administered, only a very small fraction of the dose actually hits the relevant receptors or sites of action, and most of the dose is actually wasted either by being taken up into the “wrong” tissue, removed from the “right” tissue too quickly, or destroyed on route before arrival. Scientists researching drug delivery seek to address these issues in order to maximize drug activity and minimize side effects2. Drug delivery is becoming an extremely demanding science. The reasons are essentially threefold: (a) the emergence of the more challenging low-molecular-weight molecules and bio macro molecules with either poor aqueous solubility, poor tissue permeation, or both, (b) the increased use of biological materials with poorly understood physical properties or questionable shelf life issues, and (c) the realization that if the portion of the dose responsible for adverse events could be directed away from sites where they originate, toxic side effects would become less frequent, thus benefiting the therapeutic index3,4. Microencapsulation is a useful method which prolongs the duration of drug effect significantly and improves patient compliance.
Ampicillin, a potent antibiotic with relatively short-termed stability in aqueous solutions, is used clinically to treat a broad range of bacterial infections. This drug has a shorter biological half-life (1 hrs); it needs multiple administrations in injectables and oral dosage form, which often results in dose related side effects and poor patient compliance5. Therefore, ampicillin is an ideal candidate for sustained release formulation, resulting in more reproducible drug absorption and reducing the risk of local irritations compared to single dosage forms. The aim of present work was to prolonged-release microcapsules of Ampicillin Hydrochloride (AH) were prepared by employing ethyl cellulose as a polymer in various ratios of 1:1, 1:3 & 1:4, by emulsion solvent diffusion technique.
Material and Methods
Materials
Ampicillin (Zim Labs. Pvt. Ltd., Nagpur) was used as a water insoluble model drug with a very bitter taste. Ethyl cellulose (Lobachemie. Pvt. Ltd. Mumbai) dicholoro methane (Loba chemie. Pvt. Ltd. Mumbai) acetone (Lobachemie. Pvt. Ltd., Mumbai), polyvinyl alcohol (Lobachemie. Pvt. Ltd., Mumbai) were used. All other Chemicals and solvents used were of analytical reagent grade.
Preparation of Microspheres
All microspheres were obtained by the emulsion solvent diffusion method using distilled water as an external phase, in which 1% of PVA was dissolved as an emulsifier. The internal phase consisted of a good solvent and a bridging liquid involving ampicillin polymer. At first, the drug and polymer were co-dissolved in an organic solvent mixture that was composed of ethanol, acetone (good solvent) and dichloromethane (bridging liquid). The drug solution was slowly injected via a syringe into the external water phase (poor solvent) under agitating. The system was stirred continuously for about 1 hr. Along with the good solvent diffusing into the poor solvent, droplets gradually solidified and formed microspheres6.
Then, the system was filtered to separate the microspheres from the preparation system. The resultant product was washed with distilled water and dried in an oven at 400C for 12 hr. the whole process was carried out at room temperature7.
Morphological and Topographical characterization
Microcapsules were observed with trinocular microscope (Lebo med ATC-2000, Japan) and scanning electron microscope (LEO, 435 VP, U.K.). Their diameters were determined with a pre-calibrated graduated eyepiece. Particle size was calculated by using equation.
Xg = 10 x [(ni x log Xi) / N]
Where, Xg is geometric mean diameter, ni is number of particles in range, Xi is the midpoint of range and N is the total number of particles8,9. All the experimental units were scrutinized in triplicate (n=3).
Flow Properties
Flow ability of microcapsules was investigated by determining angle of repose, bulk density,
Carr’s index and Hausner ratio. The angle of repose was determined by fixed funnel method. The microcapsules were tapped using bulk density apparatus (Electro lab tap density tester, (USP) ETP-1020) for 1000 taps in a cylinder and the change in volume was measured10-12.
Carr index and Hausner ratio were calculated by the formula:
Carr index (%) = (Df -D0) ×100 ⁄ Df
Hausner ratio = Df ⁄ D0
Where, Df is poured density; D0 is tapped density. All the experimental units were studied
in triplicate (n=3).
Drug content and encapsulation efficiency (DEE)
Accurately weighed microcapsules equivalent to 50 mg of ampicillin, were suspended in 100 mL of simulated gastric fluid (GF, phosphate buffer, pH 3.2) and kept for 24 h. Next day it was filtered after stirring and analyzed by using UV-Visible spectrophotometer (Systronic PC Based double beam Spectrophotometer-2201, Shimadzu, Japan) after suitable dilution at 320 nm. Drug entrapment efficiency (DEE) was calculated using the formula11,12.
DEE = (Practical drug content/Theoretical percent drug content) × 100
Each sample was analyzed in triplicate (n=3).
Wall thickness of microcapsules
Wall thicknesses of the microcapsules were determined by the method suggested by Luu et.al., using equation13.
h = r (1-P) d1 ⁄ 3[Pd2 + (1-P) d1]
Where, h is wall thickness; r is mean radius of microcapsules from optical microscopic observations; d1 is density of the core material; d2 is density of the coat material; p is the proportion of medicament in the microcapsules. All the test samples were examined for three times.
In vitro drug release studies of microcapsule formulations
In vitro drug release study was carried out in USP Electro Lab dissolution test apparatus using simulated gastric fluid as dissolution medium (900 mL phosphate buffer. pH 3.2, at 37±1°C, 100 rpm). An aliquot sample (5 mL) was withdrawn at an interval of 1 hr with replacement of fresh medium and analyzed for ampicillin content by UV-visible spectrophotometer at 320 nm (10, 11). All the experimental units were evaluated in triplicate (n=3). The same method was adopted for each batch of microcapsules.
Infrared spectroscopy (IR)
A Fourier Transformed Infrared Spectrophotometer (8400s, Shimadzu, Japan) was used to scan the drug samples prepared as KBr pellets14, over the range of 4000-600 cm-1.
Scanning Electron Microscopy (SEM)
The SEM analysis was carried out using a scanning electron microscope (LEO, 435 VP, U. K.). Prior to examination, samples were mounted on an aluminium stub using a double sided adhesive tape and making it electrically conductive by coating with a thin layer of gold (approximately 20 nm) in vacuum15. The scanning electron microscope was operated at an acceleration voltage of 05 KV.
Drug content uniformity
Initially, the formulations were tested for homogeneity by visual inspection. To ensure the homogeneity of drug content in the formulation of the tablet, six tubes were sampled from the different locations of the mixer and assayed for the drug content as stated above16,17. Studies were performed in triplicate for all the formulations.
Results and Discussions
All the microcapsules were found to be discrete, spherical and free flowing. The size could be separated and more uniform size of capsules could rapidly be obtained. The formulation of microcapsules has been presented in table 1. The trinocular microscopy (Figure 1) revealed that all obtained microcapsules were opaque, discrete and spherical particles with smooth surfaces, which further confirmed by SEM studies. Particle size distribution of selected microcapsules and the mean particle size for all formulations were graphically represented in figures 3. The mean diameter of the microcapsules was found to be increased with increase in proportion of coat material as shown in table 1. The particle size distribution was found as wide in formulation F3. So care must be taken during preparation of microcapsules that is stirring must be done with much high speed, there must be gradual reduction of temperature during cooling and avoidance of sticking of microcapsules18,19. The flow properties and wall thickness of the microcapsules were shown in table 1. All the formulations had excellent flow properties. The highest wall thickness (4.267 μm) belonged to F3. The wall thickness of the microcapsules mainly depended on polymer content. As usual, the wall thickness of the microcapsules increased with polymer ratio as depicted in table 1. Relatively high drug content and encapsulation efficiency were observed for each formulation presented in table 1. The increased encapsulation efficiency may be attributed to the hydrophobic nature of ethyl cellulose and ampicillin. It was found that the encapsulation efficiency improved by decreasing the polymer content. The release of drug from various formulations was from 12 hrs and more as shown in figure 2. Drug releases from the microcapsules were studied in phosphate buffer pH 7.4 for a period 12 hrs. Drug release from the microcapsules (F1 and F2) was 94-97 % in first 6 hours, with the initial burst of nearly 50% within one hour (Table 2). Drug release from microcapsules (F1 and F3) sustained the drug release up-to 12 hours20. The interaction between the drug and the carrier often leads to identifiable changes in the FT-IR profile of solid systems. FT-IR spectra at 45 scan and at a resolution of 1 cm-1 were recorded in KBr press pellets for pure drug (Figure 5A), polymer (Ethyl cellulose) (Figure 5B) and the selected (F3) microcapsule formulation (Figure 5C) of 1:4 drug / polymer ratios. In FT-IR studies, the characteristic C-N stretching at around 1620 cm-1 was clearly distinguishable in the selected formulation (F3). Additionally characteristics O-H stretching vibration at around 3250 cm-1 and N=O symmetrical and asymmetrical stretching at around 1560 and 1350 cm-1 respectively21,22, were also observed unchanged in the formulation suggesting no drug polymer chemical interaction. The morphology of the ethyl cellulose ampicillin systems prepared by thermal change method was investigated by SEM analysis (Figure 2). Microcapsules appear as small spherical particle with smooth surfaces of homogenous morphology23.
Table 1 Evaluation parameters of prepared ampicillin microcapsule
| Evaluation parameters | Formulations | ||
| F1 | F2 | F3 | |
| Particle size (μm) | 20.32±1.11 | 25.531±0.99 | 38.123±1.65 |
| Drug content (mg) | 17.032±0.23 | 11.033±0.30 | 06.425±0.25 |
| Encapsulation efficiency (%) | 84.942±1.10 | 83.421±0.76 | 84.246±0.78 |
| Bulk density (g/cc) | 1.212±0.11 | 1.357±0.28 | 1.410±0.17 |
| Angle of repose (θ) | 14.6±0.23 | 20.7±0.65 | 23.6±0.34 |
| Hausner’s ratio | 1.091±0.14 | 1.101±0.36 | 1.124±0.23 |
| Carr’s index | 07.600±0.53 | 10.760±0.24 | 07.110±0.36 |
| Wall thickness (μm) | 3.357±0.16 | 2.988±0.24 | 4.267±0.19 |
| In vitro drug release (%) | 70.258±1.10 | 68.211±1.02 | 43.334±0.84 |
F1 = drug: polymer ratio 1:1 w/w, F2 = drug: polymer ratio 1:3 w/w and F3 = drug: polymer ratio 1:4 w/w.
Table 2: In vitro release kinetics studies of DS microcapsules
| Formulation Code | Regression coefficient (r) values | ||||
| Zero order | First order | Higuchi | Hixson Crowell | Peppas | |
| F1 | 0.9625 | 0.9894 | 0.9724 | 0.9689 | 0.9365 |
| F2 | 0.9569 | 0.9874 | 0.9649 | 0.9766 | 0.9756 |
| F3 | 0.9756 | 0.9914 | 0.9847 | 0.9843 | 0.9871 |
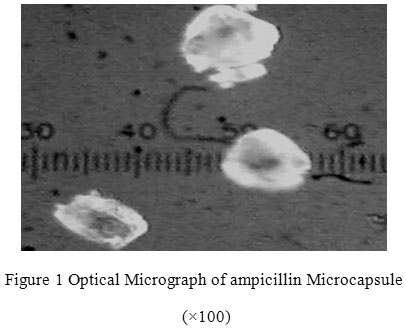 |
Figure 1: Optical Micrograph of ampicillin Microcapsule (×100).
|
 |
Figure 2: Scanning electron micrograph of ampicillin microcapsule at 05KV × 4000.
|
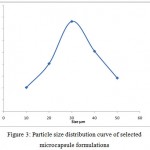 |
Figure 3: Particle size distribution curve of selected microcapsule formulations.
|
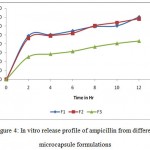 |
Figure 4: In vitro release profile of ampicillin from different microcapsule formulations.
|
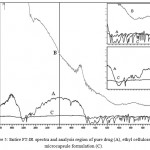 |
Figure 5: Entire FT-IR spectra and analysis region of pure drug (A), ethyl cellulose (B), microcapsule formulation (C).
|
Conclusion
In conclusion, F3, containing drug: polymer ratio 1:4, was selected as the best microcapsule. Formulation because of its slower release rate, higher entrapment efficiency and excellent flow property. F3 (containing 1% w/w of drug loaded microcapsules and 0.6% w/w of ethyl cellulose) was found to be the best because releasing almost 100% of ampicillin over a period of 36 h in simulated gastric fluid, successfully. The novel formulation design facilitated the optimization and successful development of microcapsule. It was concluded that the applied protocol can be an effective strategy for the development of safe, easy, reproducible and cost effective oral delivery systems.
Acknowledgements
The authors wish to acknowledge the University institute of pharmacy, Pt. Ravi Shankar Shukla University, Raipur (C.G.) for providing equipments and facilities needed for this work.
References
- Peppas L. B., Adv. Drug Deliv. Rev. 11, 169-176 (1993).
- Chien Y. W., Novel Drug Delivery Systems. 2nd edition, Marcel Dekker Inc, New York, 529–540 (1992).
- Karasulu H. Y., Taneri F., Sanal E., Guèneri T. and Ertan G., J. Microencapsul., 19, 357-362 (2002).
- Dandagi P. M., Manvi F. V., Gadad A. P., Mastiholimath V. S. and Patil M. B., Indian J. Pharm. Sci., 66, 631-635 (2004).
- Oliyai R. and Lindenbaum S., Int J Pharm., 73, 33-36 (1991).
- Chowdary K. P. R. and Annapurna A., East. Pharm. 9, 129-131(1992).
- Chowdary K. P. R. and Madhvi B. L. R., Pharm. Rev. 11, 137-140 (2006).
- Higuchi T., J. Pharm. Sci. 52, 1145-1149 (1963).
- Idsin B. and Lazarus J., The Theory and Practice of Industrial Pharmacy, 3rd edition Varghese Publishing House, Mumbai, 506-525 (1987).
- Bolton S., Pharmaceutical Statistics, Practical and Clinical Application. Marcel Dekker, New York, 128-39 (1997).
- Sajeev C., Vinay G., Archna R. and Saha R. N., J. Microencapsul., 19, 753-760 (2002).
- Singh J. and Robinson D. H. J. Microencapsul. 7, 67-76 (1990).
- Yamuda T., Ohnishi H. and Machida Y., J. Control. Release., 75, 271-282 (2001).
- Yazan Y., Demirel M. and Guler E., J. Microencapsul., 12, 601-607 (1995).
- Ghulam M., Ahmad M., Akhtar N., and Rasool F., Pak. J. Pharm. Sci., 22(3), 291-300 (2009).
- Alvarez-Fuente J., Fernandez-Arevalo M., Holgado M. A., Caraballo I., Lera J. M. And Rabasco A. M., Pharmazie, 49, 834–839 (1994).
- Barreto D. G., Curr. Therap. Res., 57, 79–86 (1996).
- Duchene D., Wouessidjewe D. and Ponchel G., J. Contr. Release, 62, 263–268 (1999).
- Ferrara A., Dos Santos C., Cimbro M., Gialdroni Grassi G., Int. J. Antimicrob. Agents, 7, 181–186 (1996).
- Fu X. Y. and Yang W. D., J. Hainan Med. Coll., 6, 4–6 (200).
- Gao Y., Wang Y. and Zhang L., Int. J. Pharmaceutics, 318, 62-69 (2006).
- Saini T. R., Kaushik D. and Dureja H., The Indian Pharmacist, 3(19), 72-75 (2004).
- Daharwal S. J., Saraf S., Jha A. K. and Bhusari K. P. The Indian Pharmacist, 3(19), 79-84 (2004).

This work is licensed under a Creative Commons Attribution 4.0 International License.



